

Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome
- PMID: 21177965
- PMCID: PMC3032930
- DOI: 10.1101/gr.108845.110
Multimodal RNA-seq using single-strand, double-strand, and CircLigase-based capture yields a refined and extended description of the C. elegans transcriptome
Abstract
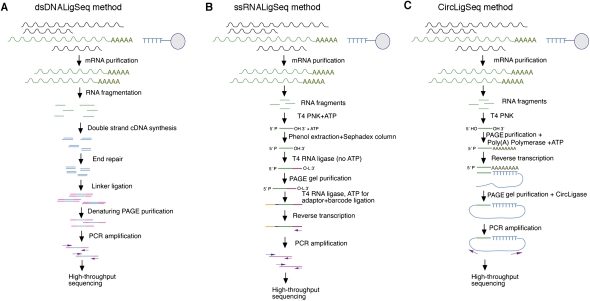
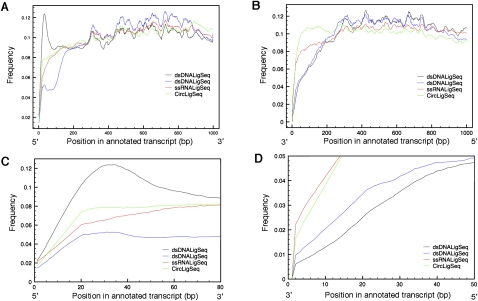
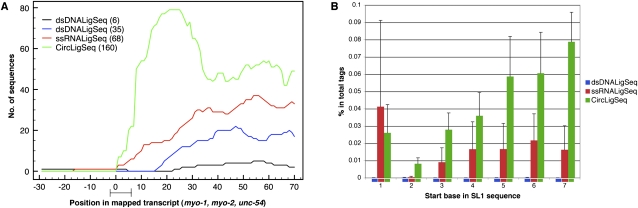
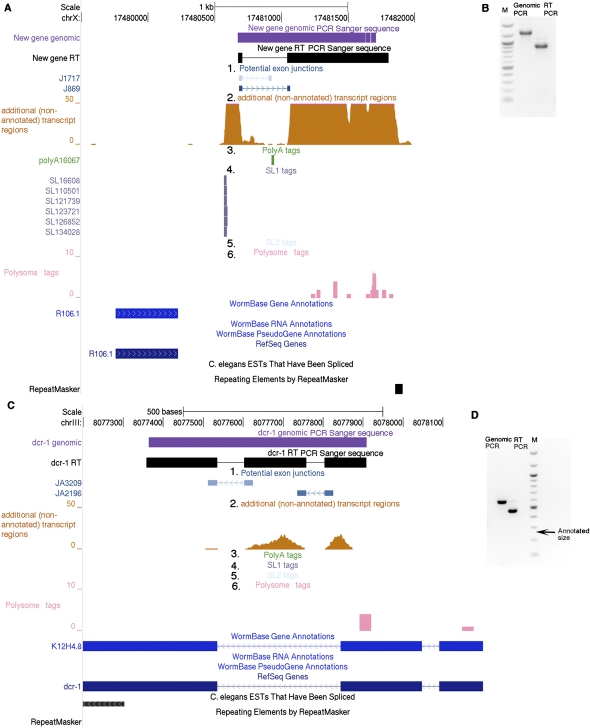
We have used a combination of three high-throughput RNA capture and sequencing methods to refine and augment the transcriptome map of a well-studied genetic model, Caenorhabditis elegans. The three methods include a standard (non-directional) library preparation protocol relying on cDNA priming and foldback that has been used in several previous studies for transcriptome characterization in this species, and two directional protocols, one involving direct capture of single-stranded RNA fragments and one involving circular-template PCR (CircLigase). We find that each RNA-seq approach shows specific limitations and biases, with the application of multiple methods providing a more complete map than was obtained from any single method. Of particular note in the analysis were substantial advantages of CircLigase-based and ssRNA-based capture for defining sequences and structures of the precise 5' ends (which were lost using the double-strand cDNA capture method). Of the three methods, ssRNA capture was most effective in defining sequences to the poly(A) junction. Using data sets from a spectrum of C. elegans strains and stages and the UCSC Genome Browser, we provide a series of tools, which facilitate rapid visualization and assignment of gene structures.
Figures
References
-
- Ambros V, Lee RC, Lavanway A, Williams PT, Jewell D 2003. MicroRNAs and other tiny endogenous RNAs in C. elegans. Curr Biol 13: 807–818 - PubMed
-
- Blumenthal T 2004. Operons in eukaryotes. Brief Funct Genomics Proteomics 3: 199–211 - PubMed
-
- Blumenthal T, Steward K 1997. RNA processing and gene structure.In C. elegans II (ed. Riddle TL et al.), pp. 117–145 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases