

Identification of functional elements and regulatory circuits by Drosophila modENCODE
- PMID: 21177974
- PMCID: PMC3192495
- DOI: 10.1126/science.1198374
Identification of functional elements and regulatory circuits by Drosophila modENCODE
Abstract
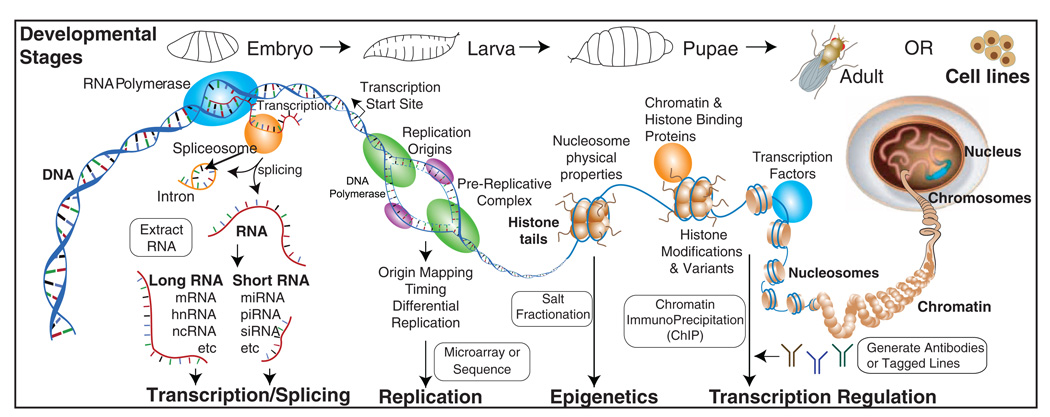
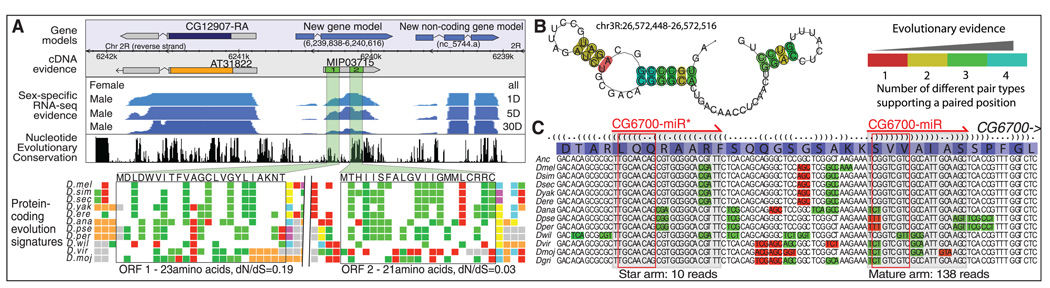
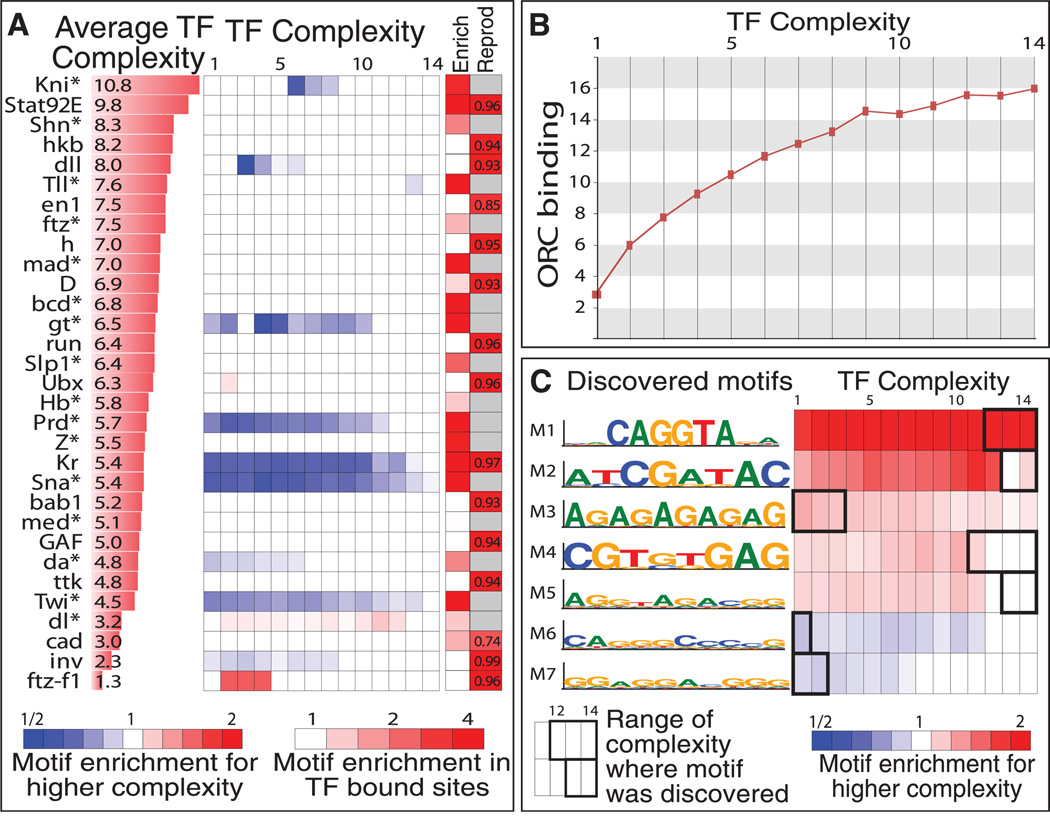
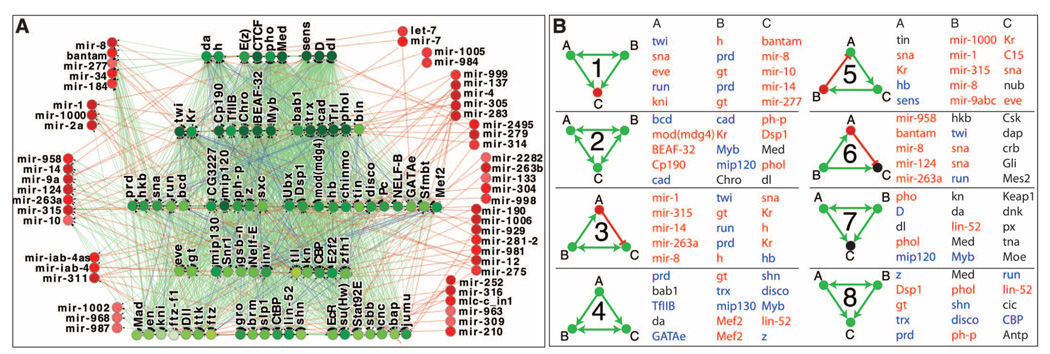
To gain insight into how genomic information is translated into cellular and developmental programs, the Drosophila model organism Encyclopedia of DNA Elements (modENCODE) project is comprehensively mapping transcripts, histone modifications, chromosomal proteins, transcription factors, replication proteins and intermediates, and nucleosome properties across a developmental time course and in multiple cell lines. We have generated more than 700 data sets and discovered protein-coding, noncoding, RNA regulatory, replication, and chromatin elements, more than tripling the annotated portion of the Drosophila genome. Correlated activity patterns of these elements reveal a functional regulatory network, which predicts putative new functions for genes, reveals stage- and tissue-specific regulators, and enables gene-expression prediction. Our results provide a foundation for directed experimental and computational studies in Drosophila and related species and also a model for systematic data integration toward comprehensive genomic and functional annotation.
Figures



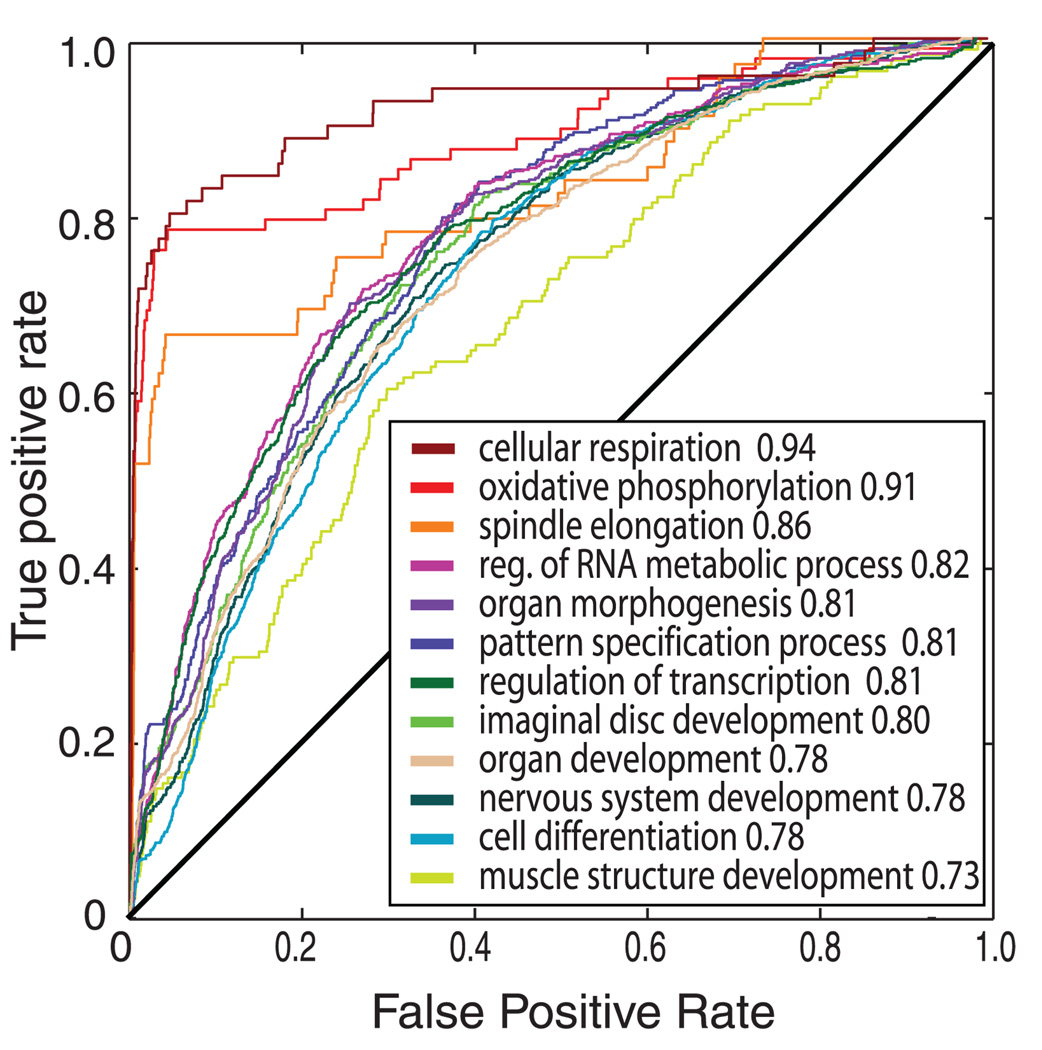
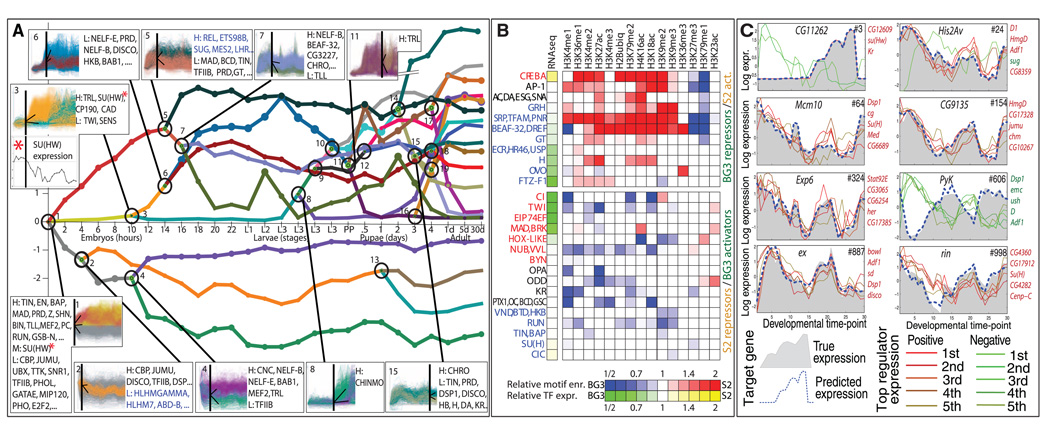
Comment in
-
Genetics. Revealing the dark matter of the genome.Science. 2010 Dec 24;330(6012):1758-9. doi: 10.1126/science.1200700. Epub 2010 Dec 22. Science. 2010. PMID: 21177977 No abstract available.
-
Functional genomics: the modENCODE guide to the genome.Nat Rev Genet. 2011 Feb;12(2):80. doi: 10.1038/nrg2942. Nat Rev Genet. 2011. PMID: 21245826 No abstract available.
-
What makes flies and worms tick.Nat Methods. 2011 Mar;8(3):204. doi: 10.1038/nmeth0311-204. Nat Methods. 2011. PMID: 21473021
References
-
-
Compared to FlyBase release 5.12 (October 2008), available at http://fb2008_09.flybase.org/
-
-
- Stapleton M, et al. Genome Biol. 2002;3 RESEARCH0080.
Publication types
MeSH terms
Substances
Grants and funding
- R01 HG004037/HG/NHGRI NIH HHS/United States
- U01HG004261/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01HG004037/HG/NHGRI NIH HHS/United States
- U01HG004279/HG/NHGRI NIH HHS/United States
- U41HG004269/HG/NHGRI NIH HHS/United States
- U01 HG004279/HG/NHGRI NIH HHS/United States
- U01HG004264/HG/NHGRI NIH HHS/United States
- R01 GM081871/GM/NIGMS NIH HHS/United States
- U01HG004274/HG/NHGRI NIH HHS/United States
- RC2HG005639/HG/NHGRI NIH HHS/United States
- ZIA DK015600/ImNIH/Intramural NIH HHS/United States
- U01HG004271/HG/NHGRI NIH HHS/United States
- U01 HG004271/HG/NHGRI NIH HHS/United States
- U01HG004258/HG/NHGRI NIH HHS/United States
- U01 HG004258/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
