

Progress in Enzyme Replacement Therapy in Glycogen Storage Disease Type II
- PMID: 21179524
- PMCID: PMC3002626
- DOI: 10.1177/1756285609103324
Progress in Enzyme Replacement Therapy in Glycogen Storage Disease Type II
Abstract
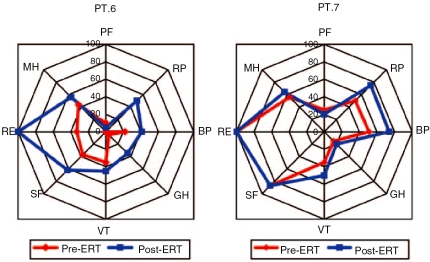
Glycogen storage disease type II (GSDII) is an autosomal recessive lysosomal disorder caused by mutations in the gene encoding alpha-glucosidase (GAA). The disease can be clinically classified into three types: a severe infantile form, a juvenile and an adultonset form. Cases with juvenile or adult onset GSDII mimic limb-girdle muscular dystrophy or polymyositis and are often characterized by respiratory involvement. GSDII patients are diagnosed by biochemical assay and by molecular characterization of the GAA gene. Ascertaining a natural history of patients with heterogeneous late-onset GSDII is useful for evaluating their progressive functional disability. A significant decline is observed over the years in skeletal and respiratory muscle function. Enzyme replacement therapy (ERT) has provided encouraging results in the infantile form. It is not yet known if ERT is effective in late-onset GSDII. We examined a series of 11 patients before and after ERT evaluating muscle strength by MRC, timed and graded functional tests, 6-minute walk test (6MWT), respiratory function by spirometric parameters and quality of life. We observed a partial improvement during a prolonged follow-up from 3 to 18 months. The use of different clinical parameters in the proposed protocol seems crucial to determine the efficacy of ERT, since not all late-onset patients respond similarly to ERT.
Keywords: glycogen storage disease type II; protocol; trial.
Figures

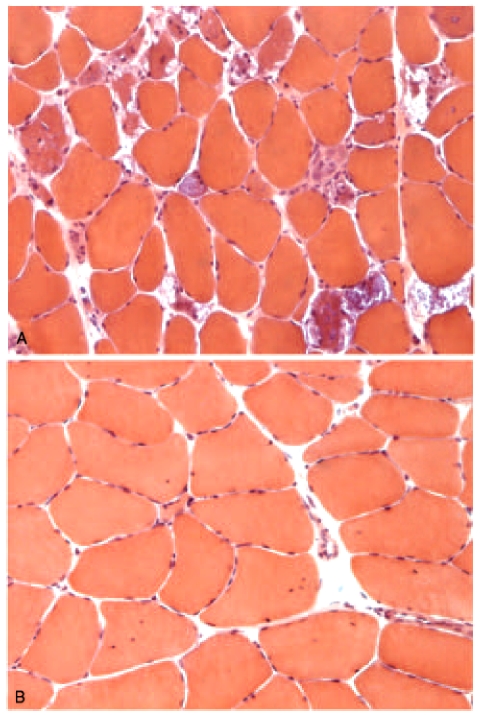
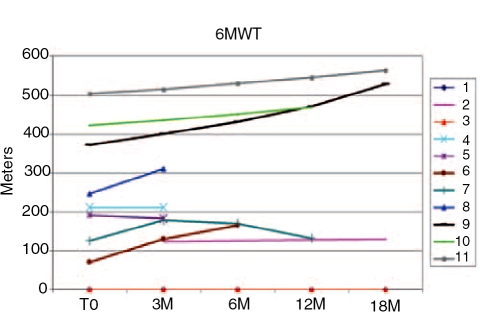
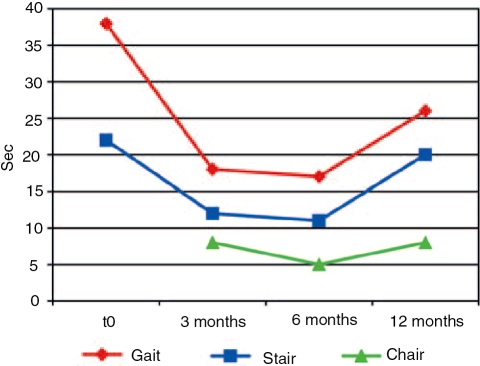
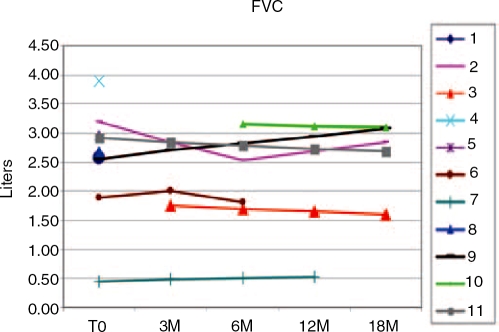
References
-
- Angelini C., Engel A.G. (1972) Comparative study of acid maltase deficiency. Biochemical differences between infantile, childhood and adult types. Arch Neurol 26: 344–349 - PubMed
-
- Angelini C., Pegoraro E., Zambito-Marsala S., Vergani L., Nascimbeni A.C., Fulizio L.et al. (2004) Adult acid maltase deficiency: an open trial with albuterol and branched-chain aminoacids. Basic Appl Myol 14: 71–78
-
- Angelini C. (2007) The role of corticosteroids in muscular dystrophy: A critical appraisal. Muscle Nerve 36: 424–435 - PubMed
-
- Angelini C., Nascimbeni A.C. (2007) Late-onset GSDII with novel GAA gene mutation. Clin Genet 71: 374–375 - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
