

Phenotypic landscape of a bacterial cell
- PMID: 21185072
- PMCID: PMC3060659
- DOI: 10.1016/j.cell.2010.11.052
Phenotypic landscape of a bacterial cell
Abstract
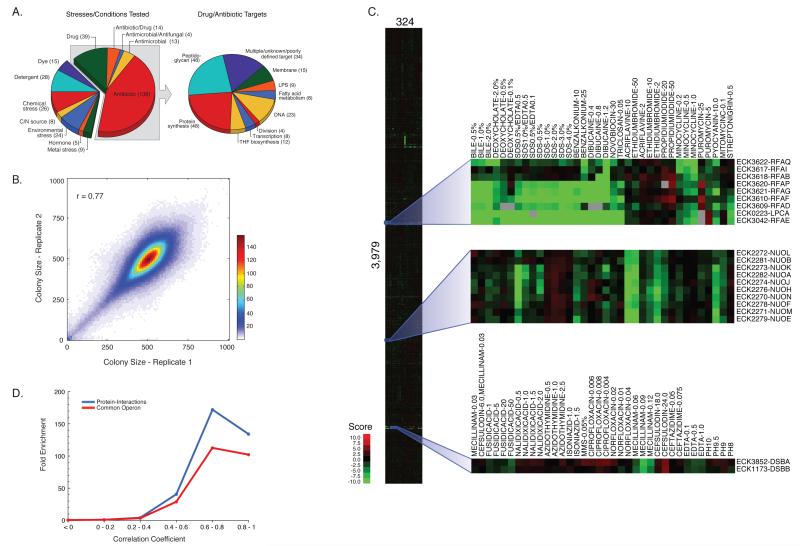
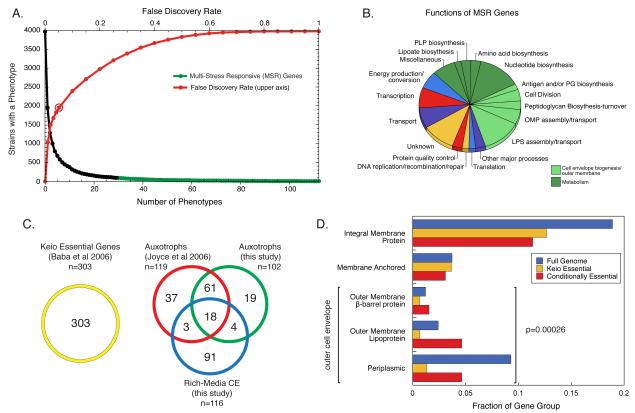
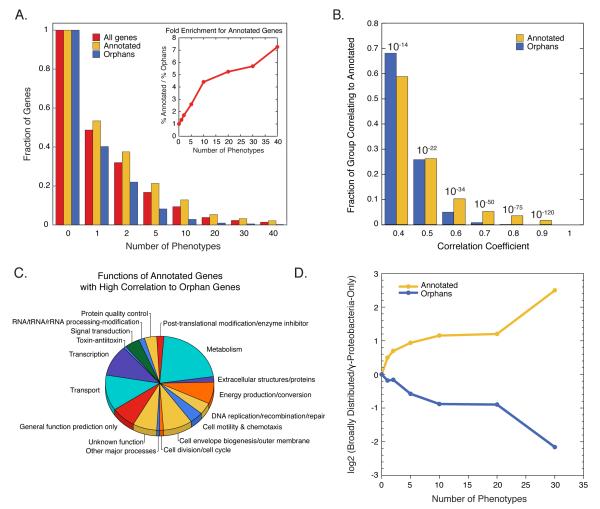
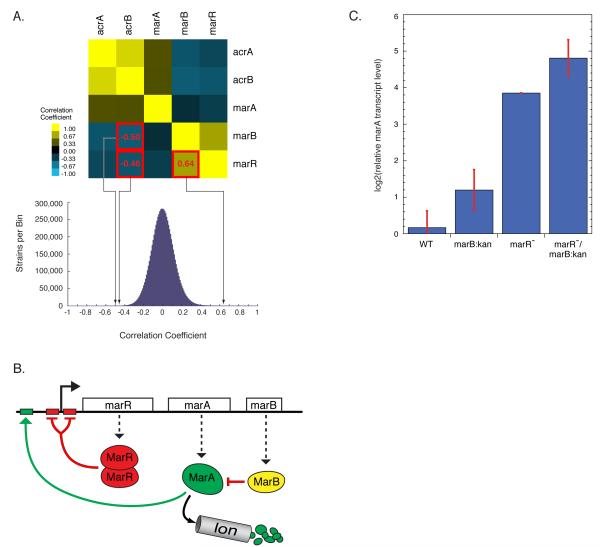
The explosion of sequence information in bacteria makes developing high-throughput, cost-effective approaches to matching genes with phenotypes imperative. Using E. coli as proof of principle, we show that combining large-scale chemical genomics with quantitative fitness measurements provides a high-quality data set rich in discovery. Probing growth profiles of a mutant library in hundreds of conditions in parallel yielded > 10,000 phenotypes that allowed us to study gene essentiality, discover leads for gene function and drug action, and understand higher-order organization of the bacterial chromosome. We highlight new information derived from the study, including insights into a gene involved in multiple antibiotic resistance and the synergy between a broadly used combinatory antibiotic therapy, trimethoprim and sulfonamides. This data set, publicly available at http://ecoliwiki.net/tools/chemgen/, is a valuable resource for both the microbiological and bioinformatic communities, as it provides high-confidence associations between hundreds of annotated and uncharacterized genes as well as inferences about the mode of action of several poorly understood drugs.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Barker CA, Farha MA, Brown ED. Chemical genomic approaches to study model microbes. Chem Biol. 2010;17:624–632. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
