

A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy
- PMID: 21186264
- PMCID: PMC4038512
- DOI: 10.1093/brain/awq294
A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy
Abstract
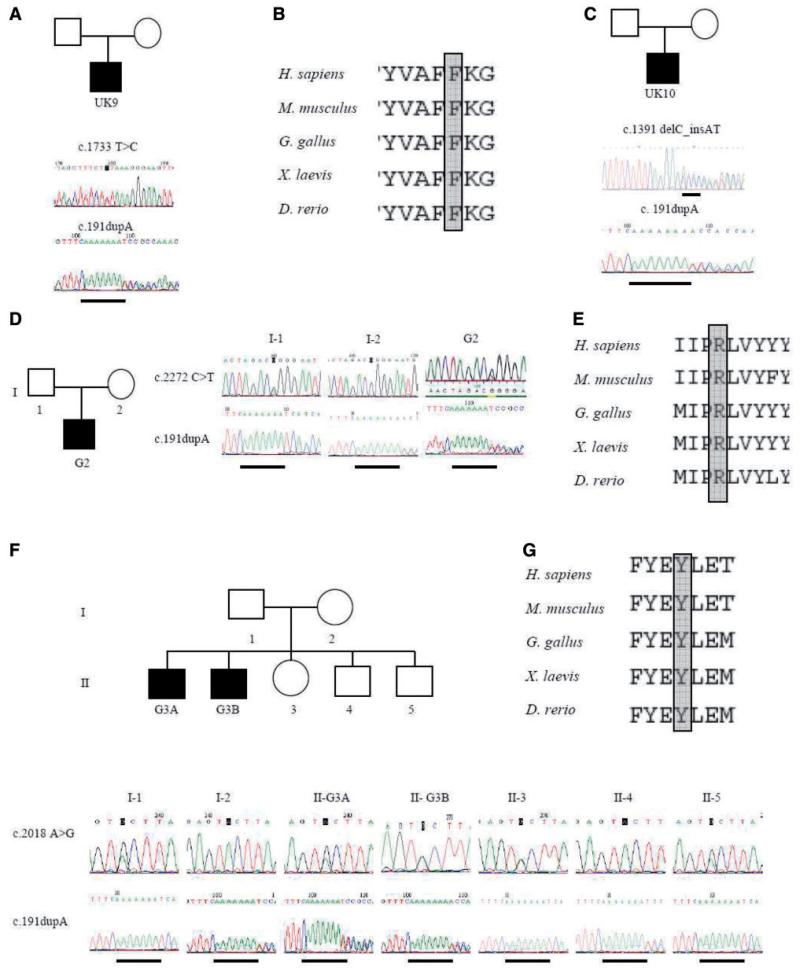
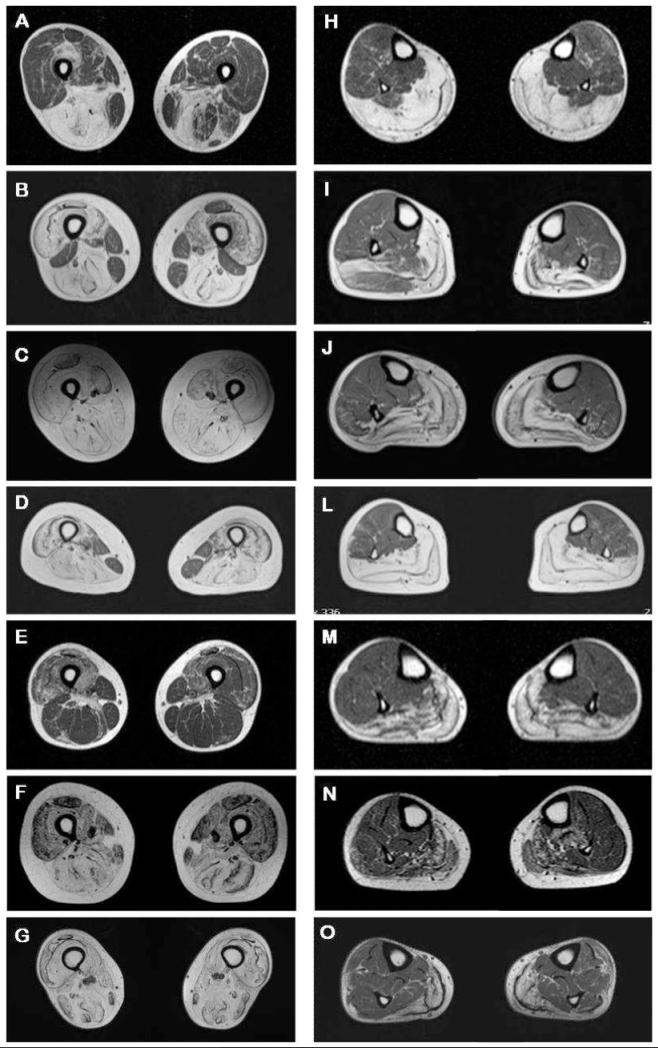
The limb-girdle muscular dystrophies are a group of disorders with wide genetic and clinical heterogeneity. Recently, mutations in the ANO5 gene, which encodes a putative calcium-activated chloride channel belonging to the Anoctamin family of proteins, were identified in five families with one of two previously identified disorders, limb-girdle muscular dystrophy 2L and non-dysferlin Miyoshi muscular dystrophy. We screened a candidate group of 64 patients from 59 British and German kindreds and found the truncating mutation, c.191dupA in exon 5 of ANO5 in 20 patients, homozygously in 15 and in compound heterozygosity with other ANO5 variants in the rest. An intragenic single nucleotide polymorphism and an extragenic microsatellite marker are in linkage disequilibrium with the mutation, suggesting a founder effect in the Northern European population. We have further defined the clinical phenotype of ANO5-associated muscular dystrophy. Patients show adult onset proximal lower limb weakness with highly raised serum creatine kinase values (average 4500 IU/l) and frequent muscle atrophy and asymmetry of muscle involvement. Onset varies from the early 20 s to 50 s and the weakness is generally slowly progressive, with most patients remaining ambulant for several decades. Distal presentation is much less common but a milder degree of distal lower limb weakness is often observed. Upper limb strength is only mildly affected and cardiac and respiratory function is normal. Females appear less frequently affected. In the North of England population we have identified eight patients with ANO5 mutations, suggesting a minimum prevalence of 0.27/100,000, twice as common as dysferlinopathy. We suggest that mutations in ANO5 represent a relatively common cause of adult onset muscular dystrophy with high serum creatine kinase and that mutation screening, particularly of the common mutation c.191dupA, should be an early step in the diagnostic algorithm of adult limb-girdle muscular dystrophy patients.
Figures



References
-
- Angelini C, Fanin M, Menegazzo E, Freda MP, Duggan DJ, Hoffman EP. Homozygous alpha-sarcoglycan mutation in two siblings: one asymptomatic and one steroid-responsive mild limb-girdle muscular dystrophy patient. Muscle Nerve. 1998;21:769–75. - PubMed
-
- Fanin M, Pegoraro E, Matsuda-Asada C, Brown RH, Jr, Angelini C. Calpain-3 and dysferlin protein screening in patients with limb-girdle dystrophy and myopathy. Neurology. 2001;56:660–5. - PubMed
-
- Guglieri M, Straub V, Bushby K, Lochmüller H. Limb-girdle muscular dystrophies. Curr Opin Neurol. 2008;21:576–84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
