

ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules
- PMID: 21187238
- PMCID: PMC4083816
- DOI: 10.1016/B978-0-12-381270-4.00019-6
ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules
Abstract
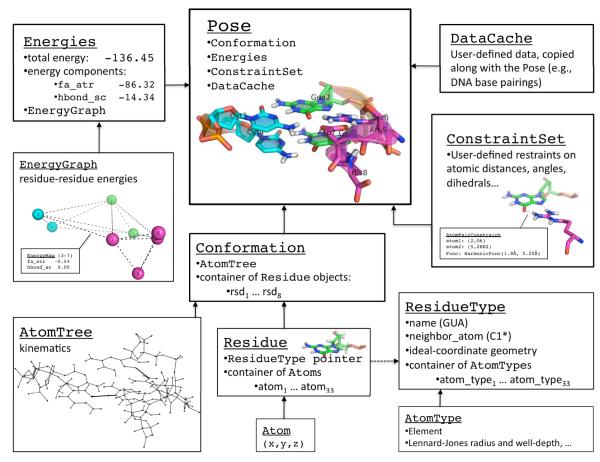
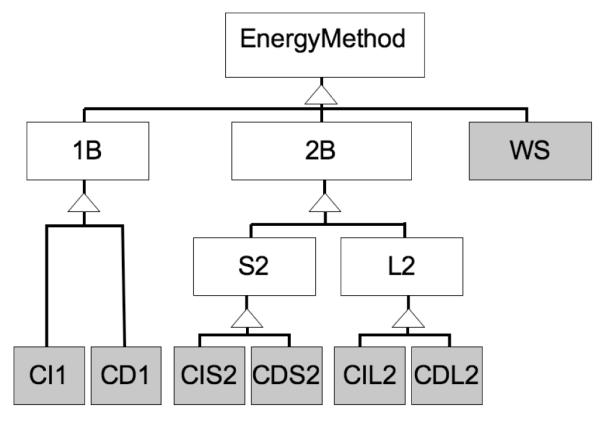
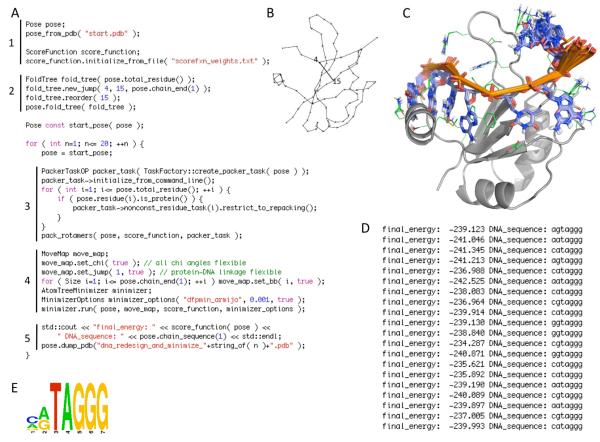
We have recently completed a full re-architecturing of the ROSETTA molecular modeling program, generalizing and expanding its existing functionality. The new architecture enables the rapid prototyping of novel protocols by providing easy-to-use interfaces to powerful tools for molecular modeling. The source code of this rearchitecturing has been released as ROSETTA3 and is freely available for academic use. At the time of its release, it contained 470,000 lines of code. Counting currently unpublished protocols at the time of this writing, the source includes 1,285,000 lines. Its rapid growth is a testament to its ease of use. This chapter describes the requirements for our new architecture, justifies the design decisions, sketches out central classes, and highlights a few of the common tasks that the new software can perform.
© 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Abe H, Braun W, Noguti T, Go N. Rapid calculation of first and second derivatives of conformational energy with respect to dihedral angles for proteins. General recurrent equations. Computers & Chemistry. 1984;8:239–247.
-
- Abagyan R, Totrov MM, Kuznetsov DN. ICM – a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994;15:488–506.
-
- Anderson DP. BOINC: A System for Public-Resource Computing and Storage. Pages 4-10 of the 5th IEEE/ACM International Conference on Grid Computing. IEEE Computer Society; Pittsburg PA: 2004.
-
- Bonneau R, Tsai J, Ruczinski I, Chivian D, Rohl C, Strauss CE, Baker D. Rosetta in CASP4: Progress in ab initio protein structure prediction. Proteins. 2001;5:119–126. - PubMed
-
- Bonneau R, Strauss CE, Rohl CA, Chivian D, Bradley P, Malmstrom L, Robertson T, Baker D. De novo prediction of three-dimensional structures for major protein families. J. Mol. Biol. 2002;322:65–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
