

Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks
- PMID: 21187424
- PMCID: PMC3021073
- DOI: 10.1073/pnas.1010954108
Nonspecific binding limits the number of proteins in a cell and shapes their interaction networks
Abstract
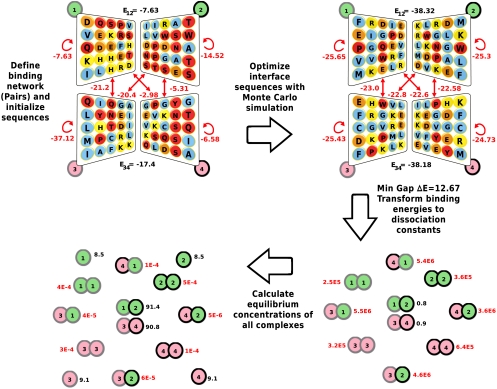
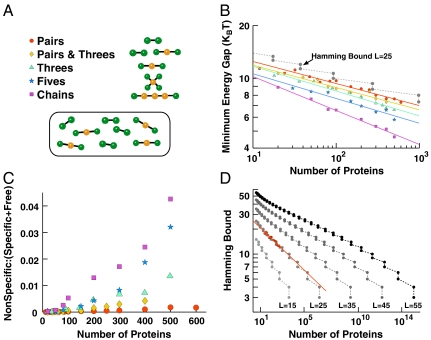
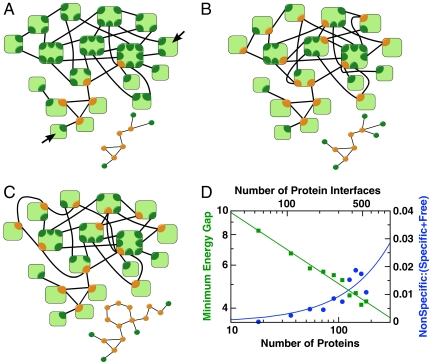
Multicellular organisms, from Caenorhabditis elegans to humans, have roughly the same number of protein encoding genes. We show that the need to prevent disease-causing nonspecific interactions between proteins provides a simple physical reason why organism complexity is not reflected in the number of distinct proteins. By collective evolution of the amino acid sequences of protein binding interfaces we estimate the degree of misbinding as a function of the number of distinct proteins. Protein interaction energies are calculated with an empirical, residue-specific energy function tuned for protein binding. We show that the achievable energy gap favoring specific over nonspecific binding decreases with protein number in a power-law fashion. From the fraction of proteins involved in nonspecific complexes as a function of increasing protein number and decreasing energy gap, we predict the limits these binding requirements place on the number of different proteins that can function effectively in a given cellular compartment. Remarkably, the optimization of binding interfaces favors networks in which a few proteins have many partners, and most proteins have few partners, consistent with a scale-free network topology. We conclude that nonspecific binding adds to the evolutionary pressure to develop scale-free protein-protein interaction networks.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Claverie J-M. What if there are only 30,000 human genes? Science. 2001;291:1255–1257. - PubMed
-
- Huang LL, Guan RJ, Pardee AB. Evolution of transcriptional control from prokaryotic beginnings to eukaryotic complexities. Crit Rev Eukaryotic Gene Expression. 1999;9:175–182. - PubMed
-
- Bird AP. Gene number, noise reduction and biological complexity. Trends Genet. 1995;11:94–100. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
