

The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time--an ultra-deep approach
- PMID: 21187908
- PMCID: PMC3002995
- DOI: 10.1371/journal.pcbi.1001022
The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time--an ultra-deep approach
Abstract
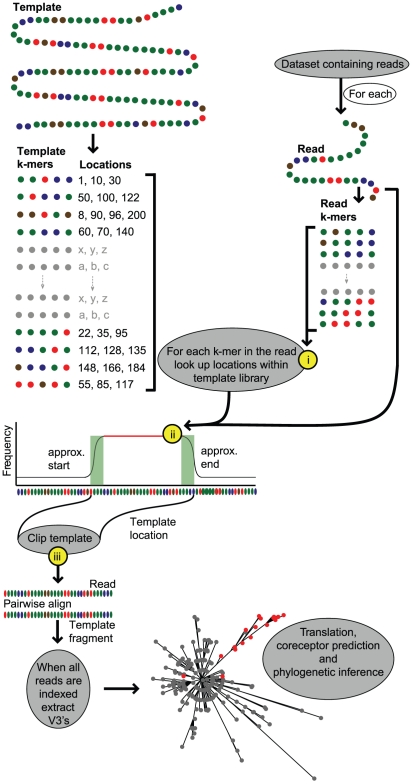
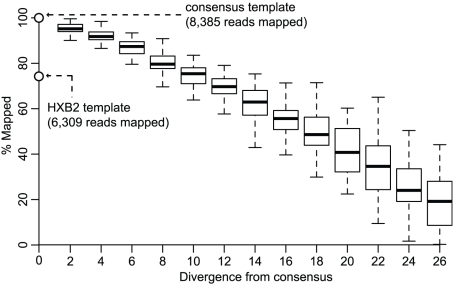
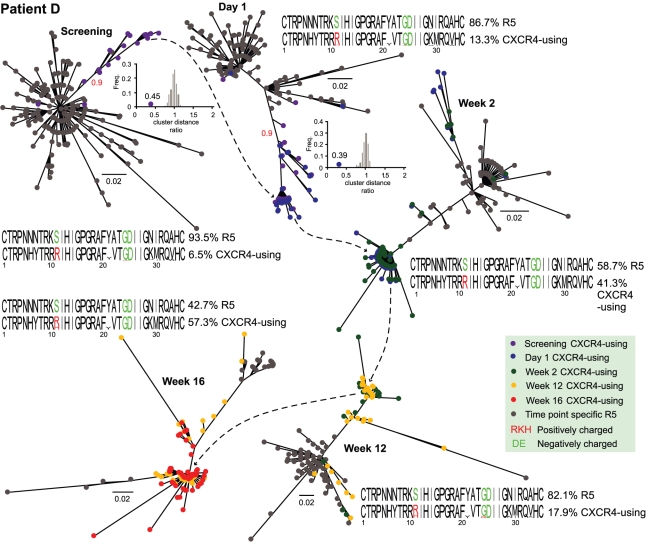
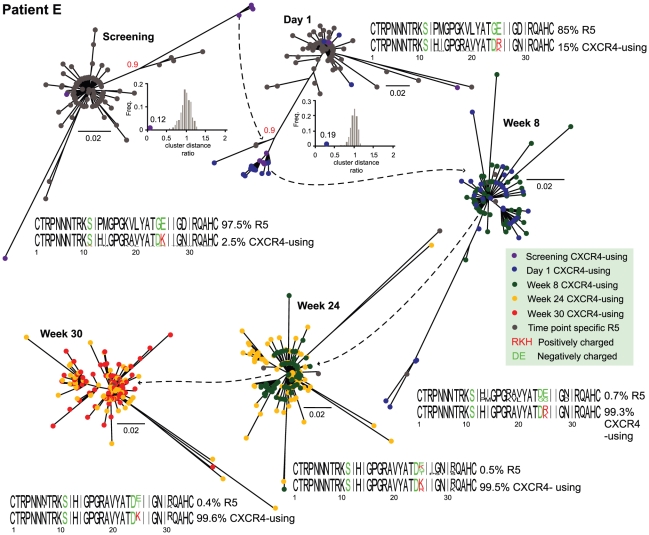
Large-scale parallel pyrosequencing produces unprecedented quantities of sequence data. However, when generated from viral populations current mapping software is inadequate for dealing with the high levels of variation present, resulting in the potential for biased data loss. In order to apply the 454 Life Sciences' pyrosequencing system to the study of viral populations, we have developed software for the processing of highly variable sequence data. Here we demonstrate our software by analyzing two temporally sampled HIV-1 intra-patient datasets from a clinical study of maraviroc. This drug binds the CCR5 coreceptor, thus preventing HIV-1 infection of the cell. The objective is to determine viral tropism (CCR5 versus CXCR4 usage) and track the evolution of minority CXCR4-using variants that may limit the response to a maraviroc-containing treatment regimen. Five time points (two prior to treatment) were available from each patient. We first quantify the effects of divergence on initial read k-mer mapping and demonstrate the importance of utilizing population-specific template sequences in relation to the analysis of next-generation sequence data. Then, in conjunction with coreceptor prediction algorithms that infer HIV tropism, our software was used to quantify the viral population structure pre- and post-treatment. In both cases, low frequency CXCR4-using variants (2.5-15%) were detected prior to treatment. Following phylogenetic inference, these variants were observed to exist as distinct lineages that were maintained through time. Our analysis, thus confirms the role of pre-existing CXCR4-using virus in the emergence of maraviroc-insensitive HIV. The software will have utility for the study of intra-host viral diversity and evolution of other fast evolving viruses, and is available from http://www.bioinf.manchester.ac.uk/segminator/.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
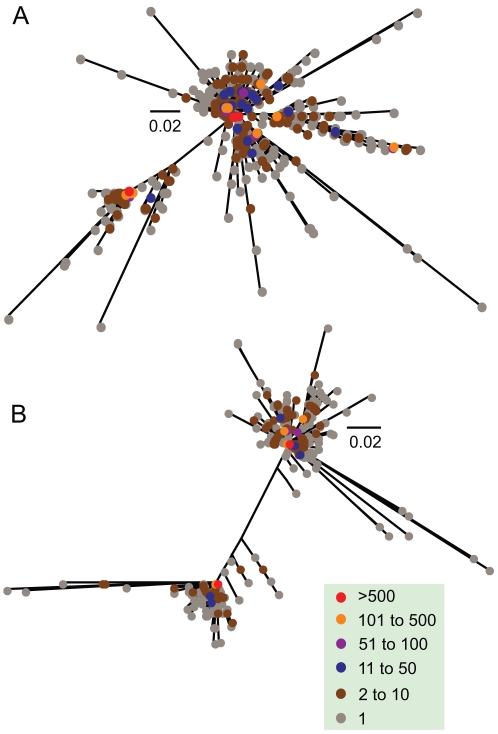
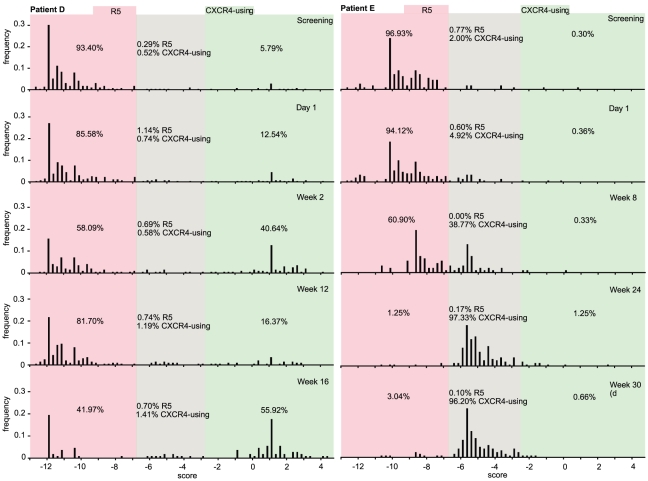
References
-
- Shendure J, Hanlee J. Next-generation DNA sequencing. Nat Biotech. 2008;26:1135–1145. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
