

Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes
- PMID: 21187929
- PMCID: PMC3004954
- DOI: 10.1371/journal.pone.0015687
Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes
Erratum in
-
Correction: Sequence Homology at the Breakpoint and Clinical Phenotype of Mitochondrial DNA Deletion Syndromes.PLoS One. 2017 Nov 20;12(11):e0188610. doi: 10.1371/journal.pone.0188610. eCollection 2017. PLoS One. 2017. PMID: 29155871 Free PMC article.
Abstract
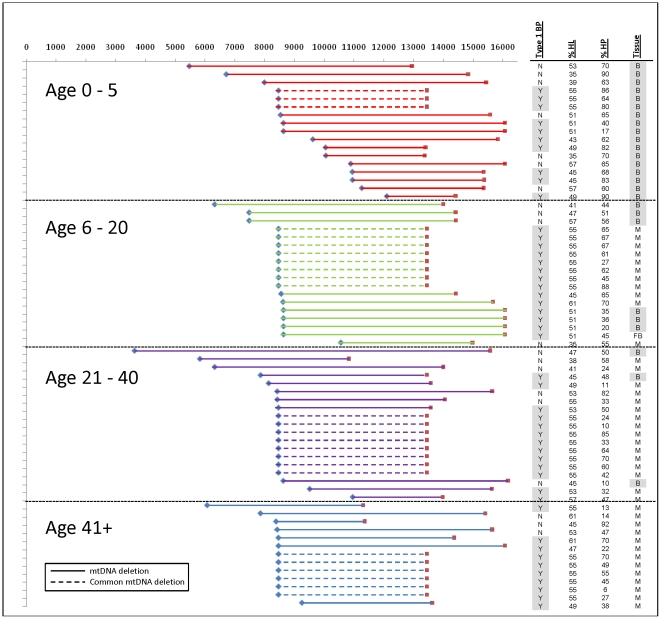
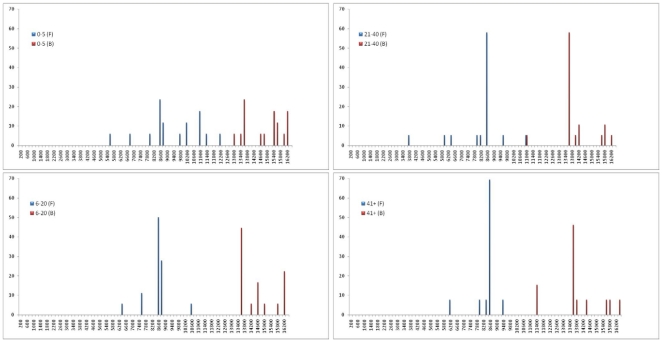
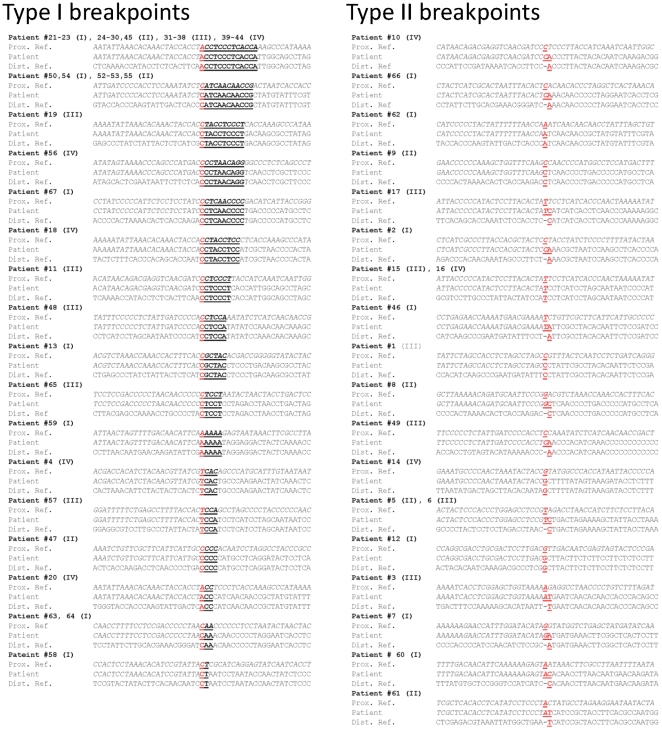
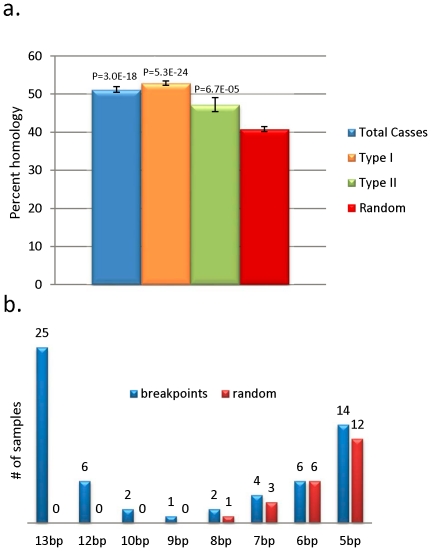
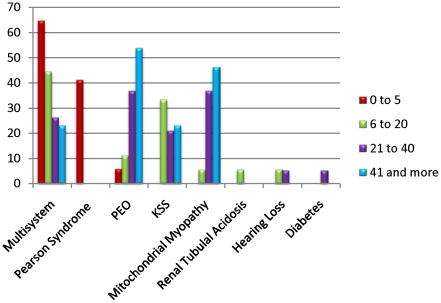
Mitochondrial DNA (mtDNA) deletions are a common cause of mitochondrial disorders. Large mtDNA deletions can lead to a broad spectrum of clinical features with different age of onset, ranging from mild mitochondrial myopathies (MM), progressive external ophthalmoplegia (PEO), and Kearns-Sayre syndrome (KSS), to severe Pearson syndrome. The aim of this study is to investigate the molecular signatures surrounding the deletion breakpoints and their association with the clinical phenotype and age at onset. MtDNA deletions in 67 patients were characterized using array comparative genomic hybridization (aCGH) followed by PCR-sequencing of the deletion junctions. Sequence homology including both perfect and imperfect short repeats flanking the deletion regions were analyzed and correlated with clinical features and patients' age group. In all age groups, there was a significant increase in sequence homology flanking the deletion compared to mtDNA background. The youngest patient group (<6 years old) showed a diffused pattern of deletion distribution in size and locations, with a significantly lower sequence homology flanking the deletion, and the highest percentage of deletion mutant heteroplasmy. The older age groups showed rather discrete pattern of deletions with 44% of all patients over 6 years old carrying the most common 5 kb mtDNA deletion, which was found mostly in muscle specimens (22/41). Only 15% (3/20) of the young patients (<6 years old) carry the 5 kb common deletion, which is usually present in blood rather than muscle. This group of patients predominantly (16 out of 17) exhibit multisystem disorder and/or Pearson syndrome, while older patients had predominantly neuromuscular manifestations including KSS, PEO, and MM. In conclusion, sequence homology at the deletion flanking regions is a consistent feature of mtDNA deletions. Decreased levels of sequence homology and increased levels of deletion mutant heteroplasmy appear to correlate with earlier onset and more severe disease with multisystem involvement.
Conflict of interest statement
Figures
References
-
- Reeve AK, Krishnan KJ, Turnbull D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann N Y Acad Sci. 2008;1147:21–29. - PubMed
-
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. - PubMed
-
- Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. 1988. Neurology. 1998;51:1525 and 1528 pages following. - PubMed
-
- Hammans SR. Mitochondrial DNA and disease. Essays Biochem. 1994;28:99–112. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
