

Regulation of endothelial function by mitochondrial reactive oxygen species
- PMID: 21194353
- PMCID: PMC3151425
- DOI: 10.1089/ars.2010.3642
Regulation of endothelial function by mitochondrial reactive oxygen species
Abstract
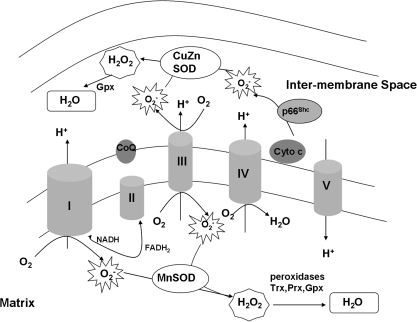
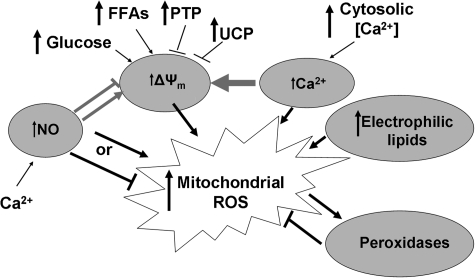
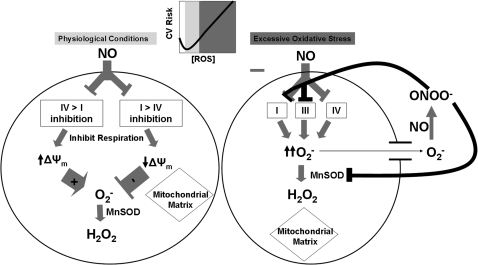
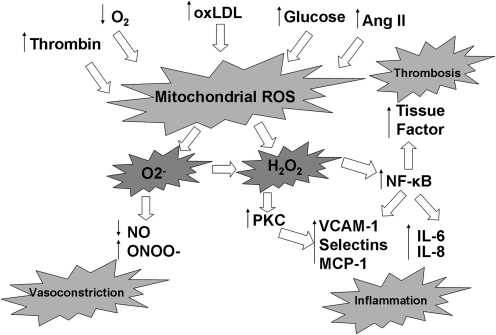
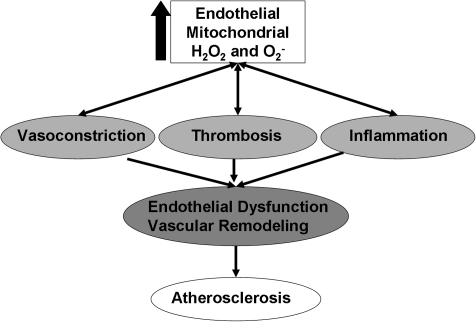
Mitochondria are well known for their central roles in ATP production, calcium homeostasis, and heme and steroid biosynthesis. However, mitochondrial reactive oxygen species (ROS), including superoxide and hydrogen peroxide, once thought to be toxic byproducts of mitochondrial physiologic activities, have recently been recognized as important cell-signaling molecules in the vascular endothelium, where their production, conversion, and destruction are highly regulated. Mitochondrial reactive oxygen species appear to regulate important vascular homeostatic functions under basal conditions in a variety of vascular beds, where, in particular, they contribute to endothelium-dependent vasodilation. On exposure to cardiovascular risk factors, endothelial mitochondria produce excessive ROS in concert with other cellular ROS sources. Mitochondrial ROS, in this setting, act as important signaling molecules activating prothrombotic and proinflammatory pathways in the vascular endothelium, a process that initially manifests itself as endothelial dysfunction and, if persistent, may lead to the development of atherosclerotic plaques. This review concentrates on emerging appreciation of the importance of mitochondrial ROS as cell-signaling molecules in the vascular endothelium under both physiologic and pathophysiologic conditions. Future potential avenues of research in this field also are discussed.
Figures
References
-
- Ali MH. Mungai PT. Schumacker PT. Stretch-induced phosphorylation of focal adhesion kinase in endothelial cells: role of mitochondrial oxidants. Am J Physiol Lung Cell Mol Physiol. 2006;291:L38–L45. - PubMed
-
- Ali MH. Pearlstein DP. Mathieu CE. Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L486–L496. - PubMed
-
- Alvarez S. Valdez LB. Zaobornyj T. Boveris A. Oxygen dependence of mitochondrial nitric oxide synthase activity. Biochem Biophys Res Commun. 2003;305:771–775. - PubMed
-
- Anderson S. Bankier AT. Barrell BG. de Bruijn MH. Coulson AR. Drouin J. Eperon IC. Nierlich DP. Roe BA. Sanger F. Schreier PH. Smith AJ. Staden R. Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
