

Layers of epistasis: genome-wide regulatory networks and network approaches to genome-wide association studies
- PMID: 21197657
- PMCID: PMC3062944
- DOI: 10.1002/wsbm.132
Layers of epistasis: genome-wide regulatory networks and network approaches to genome-wide association studies
Abstract
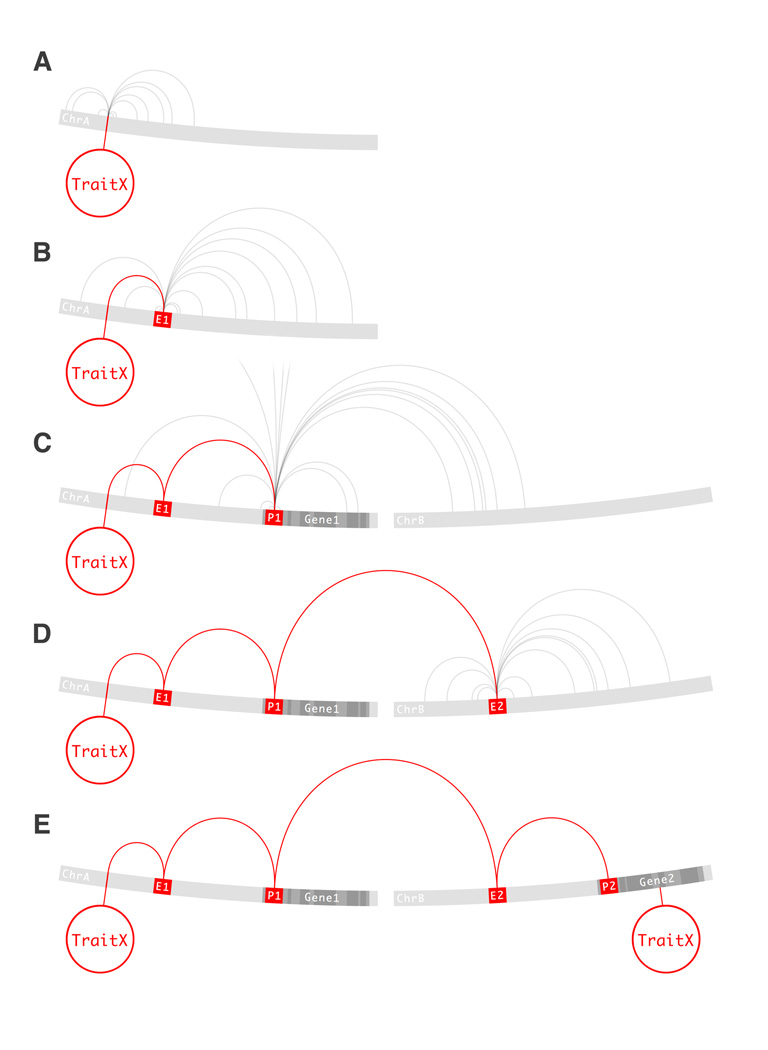
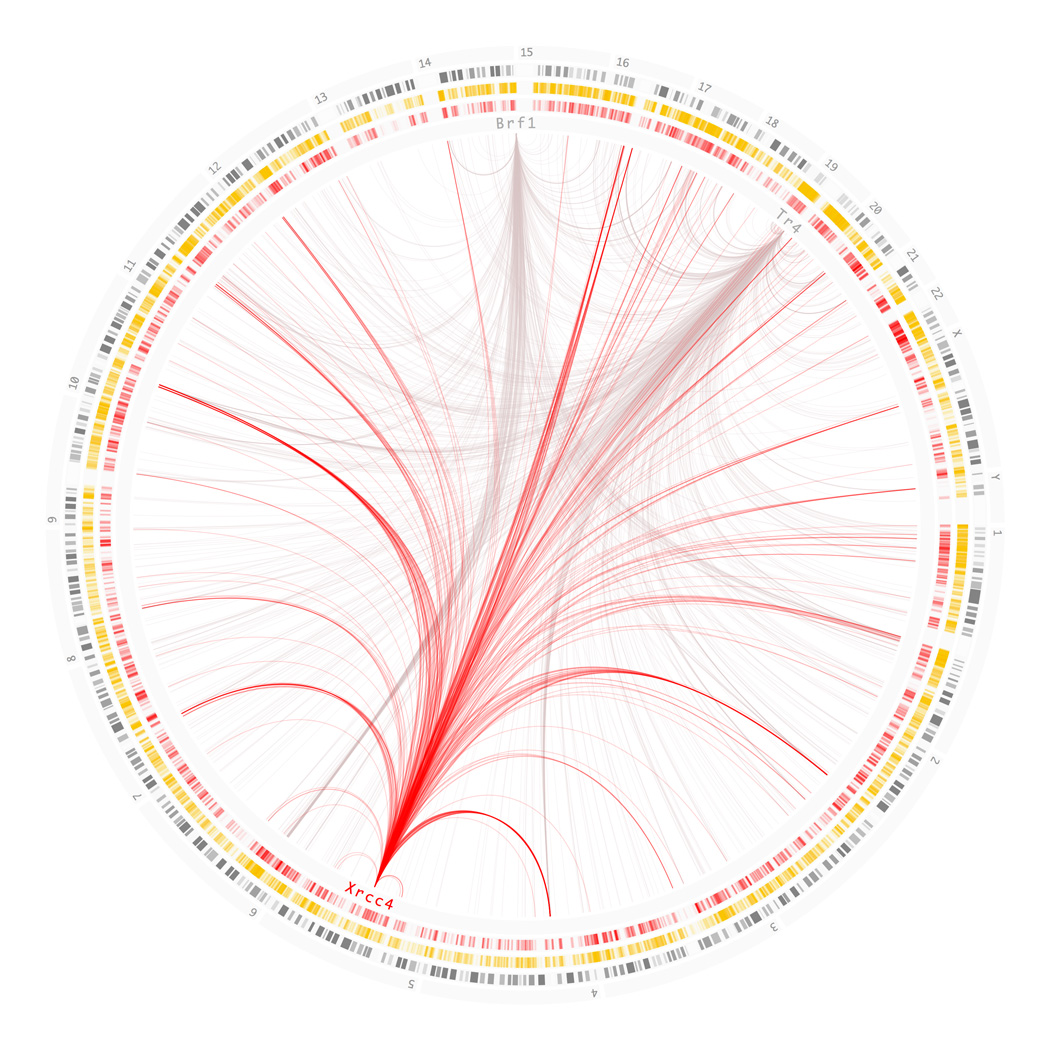
The conceptual foundation of the genome-wide association study (GWAS) has advanced unchecked since its conception. A revision might seem premature as the potential of GWAS has not been fully realized. Multiple technical and practical limitations need to be overcome before GWAS can be fairly criticized. But with the completion of hundreds of studies and a deeper understanding of the genetic architecture of disease, warnings are being raised. The results compiled to date indicate that risk-associated variants lie predominantly in noncoding regions of the genome. Additionally, alternative methodologies are uncovering large and heterogeneous sets of rare variants underlying disease. The fear is that, even in its fulfillment, the current GWAS paradigm might be incapable of dissecting all kinds of phenotypes. In the following text, we review several initiatives that aim to overcome these limitations. The overarching theme of these studies is the inclusion of biological knowledge to both the analysis and interpretation of genotyping data. GWAS is uninformed of biology by design and although there is some virtue in its simplicity, it is also its most conspicuous deficiency. We propose a framework in which to integrate these novel approaches, both empirical and theoretical, in the form of a genome-wide regulatory network (GWRN). By processing experimental data into networks, emerging data types based on chromatin immunoprecipitation are made computationally tractable. This will give GWAS re-analysis efforts the most current and relevant substrates, and root them firmly on our knowledge of human disease.
Copyright © 2010 John Wiley & Sons, Inc.
Figures
Similar articles
-
A whole-genome simulator capable of modeling high-order epistasis for complex disease.Genet Epidemiol. 2013 Nov;37(7):686-94. doi: 10.1002/gepi.21761. Epub 2013 Oct 1. Genet Epidemiol. 2013. PMID: 24114848 Free PMC article.
-
Gene-set association and epistatic analyses reveal complex gene interaction networks affecting flowering time in a worldwide barley collection.J Exp Bot. 2019 Oct 24;70(20):5603-5616. doi: 10.1093/jxb/erz332. J Exp Bot. 2019. PMID: 31504706 Free PMC article.
-
iLOCi: a SNP interaction prioritization technique for detecting epistasis in genome-wide association studies.BMC Genomics. 2012;13 Suppl 7(Suppl 7):S2. doi: 10.1186/1471-2164-13-S7-S2. Epub 2012 Dec 13. BMC Genomics. 2012. PMID: 23281813 Free PMC article.
-
Full genetic analysis for genome-wide association study of Fangji: a powerful approach for effectively dissecting the molecular architecture of personalized traditional Chinese medicine.Acta Pharmacol Sin. 2018 Jun;39(6):906-911. doi: 10.1038/aps.2017.137. Epub 2018 Feb 8. Acta Pharmacol Sin. 2018. PMID: 29417942 Free PMC article. Review.
-
Post genome-wide association analysis: dissecting computational pathway/network-based approaches.Brief Bioinform. 2019 Mar 25;20(2):690-700. doi: 10.1093/bib/bby035. Brief Bioinform. 2019. PMID: 29701762 Free PMC article. Review.
Cited by
-
Pathway-based genetic analysis of preterm birth.Genomics. 2013 Mar;101(3):163-70. doi: 10.1016/j.ygeno.2012.12.005. Epub 2013 Jan 6. Genomics. 2013. PMID: 23298525 Free PMC article.
-
Genomic analyses with biofilter 2.0: knowledge driven filtering, annotation, and model development.BioData Min. 2013 Dec 30;6(1):25. doi: 10.1186/1756-0381-6-25. BioData Min. 2013. PMID: 24378202 Free PMC article.
-
Grid-based stochastic search for hierarchical gene-gene interactions in population-based genetic studies of common human diseases.BioData Min. 2017 May 30;10:19. doi: 10.1186/s13040-017-0139-3. eCollection 2017. BioData Min. 2017. PMID: 28572842 Free PMC article.
-
Analysis of gene-gene interactions.Curr Protoc Hum Genet. 2011 Jul;Chapter 1:Unit1.14. doi: 10.1002/0471142905.hg0114s70. Curr Protoc Hum Genet. 2011. PMID: 21735376 Free PMC article.
-
Structure and dynamics of molecular networks: a novel paradigm of drug discovery: a comprehensive review.Pharmacol Ther. 2013 Jun;138(3):333-408. doi: 10.1016/j.pharmthera.2013.01.016. Epub 2013 Feb 4. Pharmacol Ther. 2013. PMID: 23384594 Free PMC article. Review.
References
-
- Andrew AS, Nelson HH, Kelsey KT, Moore JH, Meng AC, Casella DP, Tosteson TD, Schned AR, Karagas MR. Concordance of multiple analytical approaches demonstrates a complex relationship between dna repair gene snps, smoking and bladder cancer susceptibility. Carcinogenesis. 2006;27(5):1030–1037. - PubMed
-
- Askland K, Read C, Moore J. Pathways-based analyses of whole-genome association study data in bipolar disorder reveal genes mediating ion channel activity and synaptic neurotransmission. Hum Genet. 2009;125(1):63–79. - PubMed
-
- Bobadilla JL, Macek M, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of cftr mutations–correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. - PubMed
-
- Bucan M, Abrahams BS, Wang K, Glessner JT, Herman EI, Sonnenblick LI, Retuerto AIA, Imielinski M, Hadley D, Bradfield JP, Kim C, Gidaya NB, Lindquist I, Hutman T, Sigman M, Kustanovich V, Lajonchere CM, Singleton A, Kim J, Wassink TH, McMahon WM, Owley T, Sweeney JA, Coon H, Nurnberger JI, Li M, Cantor RM, Minshew NJ, Sutcliffe JS, Cook EH, Dawson G, Buxbaum JD, Grant SFA, Schellenberg GD, Geschwind DH, Hakonarson H. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 2009;5(6):e1000536. - PMC - PubMed
-
- Chan CS, Song JS. Ccctc-binding factor confines the distal action of estrogen receptor. Cancer Research. 2008;68(21):9041–9049. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
