

Glucocorticosteroids: current and future directions
- PMID: 21198556
- PMCID: PMC3085866
- DOI: 10.1111/j.1476-5381.2010.01199.x
Glucocorticosteroids: current and future directions
Abstract
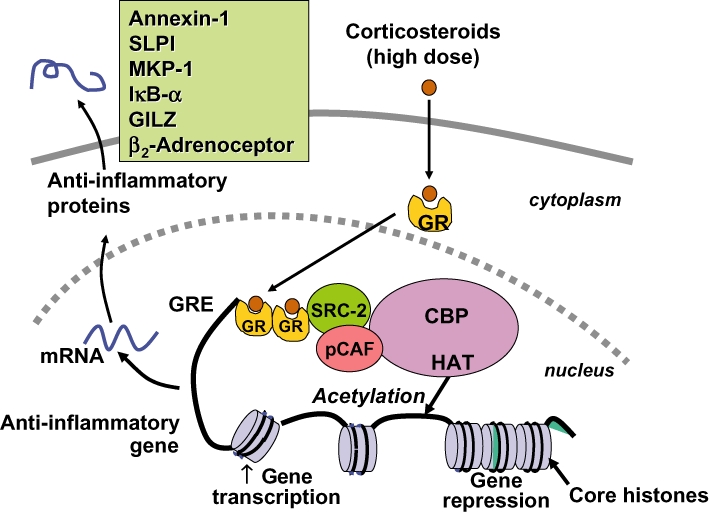
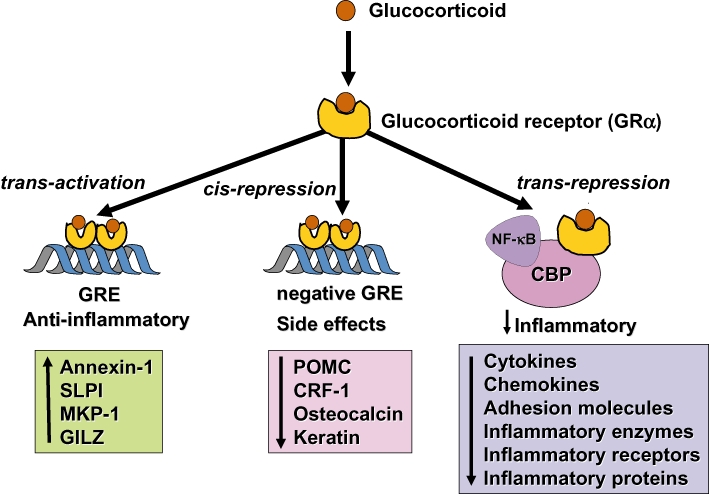
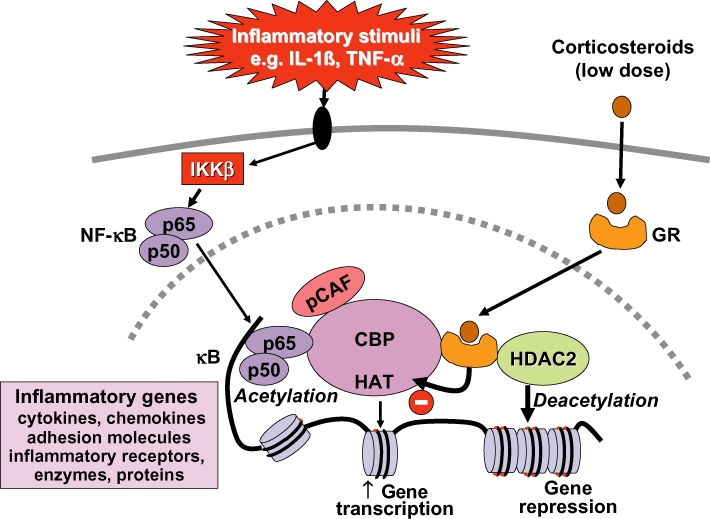
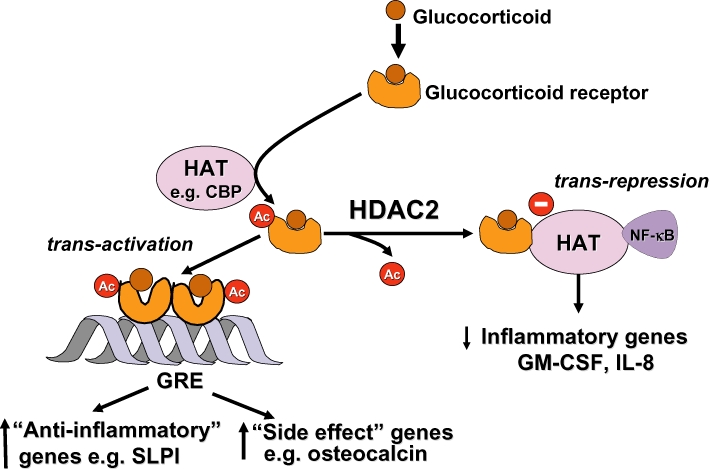
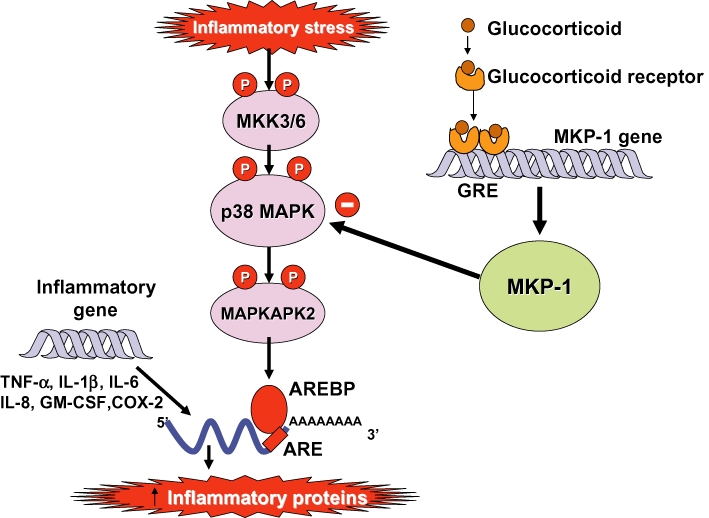
Glucocorticoids are the most effective anti-inflammatory therapy for asthma yet are relatively ineffective in chronic obstructive pulmonary disease. Glucocorticoids suppress inflammation via several molecular mechanisms. Glucocorticoids suppress the multiple inflammatory genes that are activated in chronic inflammatory diseases, such as asthma, by reversing histone acetylation of activated inflammatory genes through binding of ligand-bound glucocorticoid receptors (GR) to co-activator molecules and recruitment of histone deacetylase-2 to the activated inflammatory gene transcription complex (trans-repression). At higher concentrations of glucocorticoids GR homodimers interact with DNA recognition sites to activate transcription through increased histone acetylation of anti-inflammatory genes and transcription of several genes linked to glucocorticoid side effects (trans-activation). Glucocorticoids also have post-transcriptional effects and decrease stability of some pro-inflammatory mRNA species. Decreased glucocorticoid responsiveness is found in patients with severe asthma and asthmatics who smoke, as well as in all patients with chronic obstructive pulmonary disease. Several molecular mechanisms of glucocorticoid resistance have now been identified which involve post-translational modifications of GR. Histone deacetylase-2 is markedly reduced in activity and expression as a result of oxidative/nitrative stress so that inflammation becomes resistant to the anti-inflammatory actions of glucocorticoids. Dissociated glucocorticoids and selective GR modulators which show improved trans-repression over trans-activation effects have been developed to reduce side effects, but so far it has been difficult to dissociate anti-inflammatory effects from adverse effects. In patients with glucocorticoid resistance alternative anti-inflammatory treatments are being investigated as well as drugs that may reverse the molecular mechanisms of glucocorticoid resistance.
© 2011 The Author. British Journal of Pharmacology © 2011 The British Pharmacological Society.
Figures
References
-
- Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest. 2008;134:394–401. - PubMed
-
- Ahmad T, Barnes PJ, Adcock IM. Overcoming steroid insensitivity in smoking asthmatics. Curr Opin Investig Drugs. 2008;9:470–477. - PubMed
-
- van den Akker EL, Russcher H, van Rossum EF, Brinkmann AO, de Jong FH, Hokken A, et al. Glucocorticoid receptor polymorphism affects transrepression but not transactivation. J Clin Endocrinol Metab. 2006;91:2800–2803. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
