

Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes
- PMID: 21199327
- PMCID: PMC3822958
- DOI: 10.1111/j.1582-4934.2010.01249.x
Interplay between Ca2+ cycling and mitochondrial permeability transition pores promotes reperfusion-induced injury of cardiac myocytes
Abstract
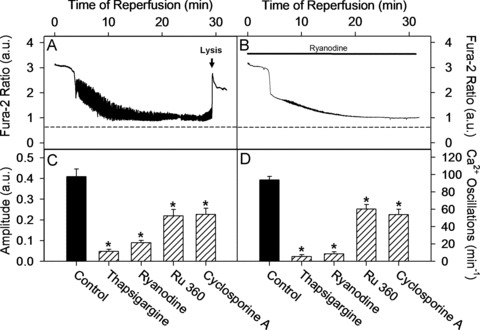
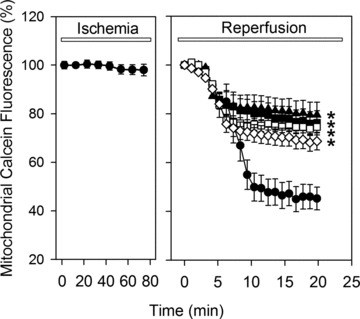
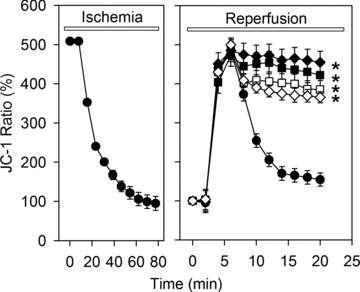
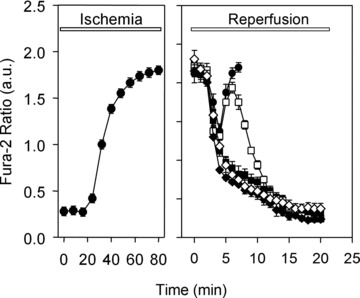
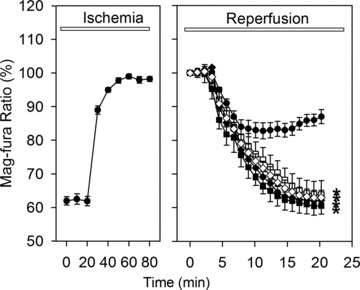
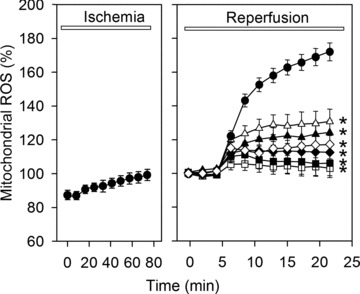
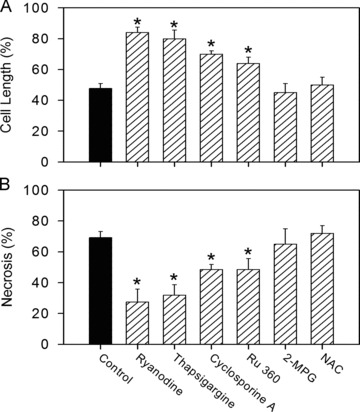
Uncontrolled release of Ca(2+) from the sarcoplasmic reticulum (SR) contributes to the reperfusion-induced cardiomyocyte injury, e.g. hypercontracture and necrosis. To find out the underlying cellular mechanisms of this phenomenon, we investigated whether the opening of mitochondrial permeability transition pores (MPTP), resulting in ATP depletion and reactive oxygen species (ROS) formation, may be involved. For this purpose, isolated cardiac myocytes from adult rats were subjected to simulated ischemia and reperfusion. MPTP opening was detected by calcein release and by monitoring the ΔΨ(m). Fura-2 was used to monitor cytosolic [Ca(2+)](i) or mitochondrial calcium [Ca(2+)](m), after quenching the cytosolic compartment with MnCl(2). Mitochondrial ROS [ROS](m) production was detected with MitoSOX Red and mag-fura-2 was used to monitor Mg(2+) concentration, which reflects changes in cellular ATP. Necrosis was determined by propidium iodide staining. Reperfusion led to a calcein release from mitochondria, ΔΨ(m) collapse and disturbance of ATP recovery. Simultaneously, Ca(2+) oscillations occurred, [Ca(2+)](m) and [ROS](m) increased, cells developed hypercontracture and underwent necrosis. Inhibition of the SR-driven Ca(2+) cycling with thapsigargine or ryanodine prevented mitochondrial dysfunction, ROS formation and MPTP opening. Suppression of the mitochondrial Ca(2+) uptake (Ru360) or MPTP (cyclosporine A) significantly attenuated Ca(2+) cycling, hypercontracture and necrosis. ROS scavengers (2-mercaptopropionyl glycine or N-acetylcysteine) had no effect on these parameters, but reduced [ROS](m). In conclusion, MPTP opening occurs early during reperfusion and is due to the Ca(2+) oscillations originating primarily from the SR and supported by MPTP. The interplay between Ca(2+) cycling and MPTP promotes the reperfusion-induced cardiomyocyte hypercontracture and necrosis. Mitochondrial ROS formation is a result rather than a cause of MPTP opening.
© 2011 The Authors Journal of Cellular and Molecular Medicine © 2011 Foundation for Cellular and Molecular Medicine/Blackwell Publishing Ltd.
Figures
References
-
- Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion–a target for cardioprotection. Cardiovasc Res. 2004;61:372–5. - PubMed
-
- Hausenloy DJ, Duchen MR, Yellon DM. Inhibiting mitochondrial permeability transition pore opening at reperfusion protects against ischaemia-reperfusion injury. Cardiovasc Res. 2003;60:617–25. - PubMed
-
- Nakagawa T, Shimizu S, Watanabe T, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8. - PubMed
-
- Piot C, Croisille P, Staat P, et al. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med. 2008;359:473–81. - PubMed
-
- Halestrap AP. What is the mitochondrial permeability transition pore. J Mol Cell Cardiol. 2009;46:821–31. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
