

Over-expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy
- PMID: 21199860
- PMCID: PMC3043663
- DOI: 10.1093/hmg/ddq559
Over-expression of BCL2 rescues muscle weakness in a mouse model of oculopharyngeal muscular dystrophy
Abstract
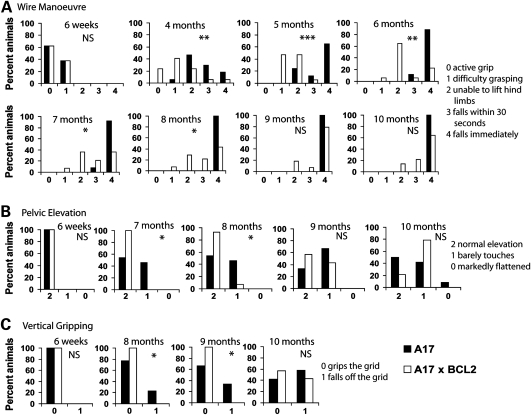
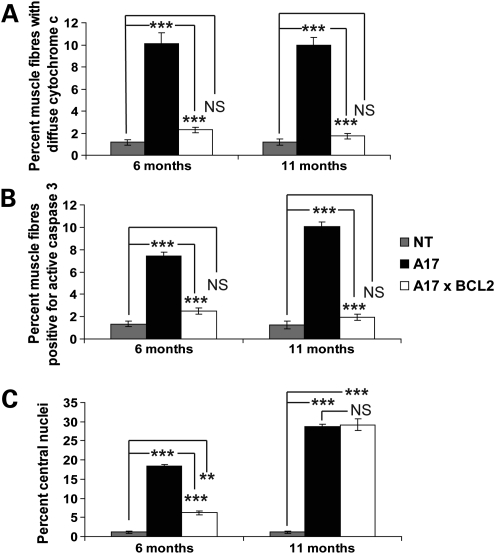
Oculopharyngeal muscular dystrophy (OPMD) is a late-onset muscular dystrophy caused by a polyalanine expansion mutation in the coding region of the poly-(A) binding protein nuclear 1 (PABPN1) gene. In unaffected individuals, (GCG)(6) encodes the first 6 alanines in a homopolymeric stretch of 10 alanines. In most patients, this (GCG)(6) repeat is expanded to (GCG)(8-13), leading to a stretch of 12-17 alanines in mutant PABPN1, which is thought to confer a toxic gain of function. Thus, OPMD has been modelled by expressing mutant PABPN1 transgenes in the presence of endogenous copies of the gene in cells and mice. In these models, increased apoptosis is seen, but it is unclear whether this process mediates OPMD. The role of apoptosis in the pathogenesis of different muscular dystrophies is unclear. Blocking apoptosis ameliorates muscle disease in some mouse models of muscular dystrophy such as laminin α-2-deficient mice, but not in others such as dystrophin-deficient (mdx) mice. Here we demonstrate that apoptosis is not only involved in the pathology of OPMD but also is a major contributor to the muscle weakness and dysfunction in this disease. Genetically blocking apoptosis by over-expressing BCL2 ameliorates muscle weakness in our mouse model of OPMD (A17 mice). The effect of BCL2 co-expression on muscle weakness is transient, since muscle weakness is apparent in mice expressing both A17 and BCL2 transgenes at late time points. Thus, while apoptosis is a major pathway that causes muscle weakness in OPMD, other cell death pathways may also contribute to the disease when apoptosis is inhibited.
Figures




Similar articles
-
Doxycycline attenuates and delays toxicity of the oculopharyngeal muscular dystrophy mutation in transgenic mice.Nat Med. 2005 Jun;11(6):672-7. doi: 10.1038/nm1242. Epub 2005 May 1. Nat Med. 2005. PMID: 15864313
-
Novel mouse models of oculopharyngeal muscular dystrophy (OPMD) reveal early onset mitochondrial defects and suggest loss of PABPN1 may contribute to pathology.Hum Mol Genet. 2017 Sep 1;26(17):3235-3252. doi: 10.1093/hmg/ddx206. Hum Mol Genet. 2017. PMID: 28575395 Free PMC article.
-
Mitochondrial localization of PABPN1 in oculopharyngeal muscular dystrophy.Lab Invest. 2019 Nov;99(11):1728-1740. doi: 10.1038/s41374-019-0243-8. Epub 2019 Mar 20. Lab Invest. 2019. PMID: 30894671
-
Progress in understanding the pathogenesis of oculopharyngeal muscular dystrophy.Can J Neurol Sci. 2003 Feb;30(1):8-14. doi: 10.1017/s0317167100002365. Can J Neurol Sci. 2003. PMID: 12619777 Review.
-
Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies.Biochim Biophys Acta. 2007 Feb;1772(2):173-85. doi: 10.1016/j.bbadis.2006.10.003. Epub 2006 Oct 11. Biochim Biophys Acta. 2007. PMID: 17110089 Review.
Cited by
-
Systemic Delivery of a Monoclonal Antibody to Immunologically Block Myostatin in the A17 Mouse Model of OPMD.Methods Mol Biol. 2023;2587:557-568. doi: 10.1007/978-1-0716-2772-3_30. Methods Mol Biol. 2023. PMID: 36401050
-
Apoptotic cell death in disease-Current understanding of the NCCD 2023.Cell Death Differ. 2023 May;30(5):1097-1154. doi: 10.1038/s41418-023-01153-w. Epub 2023 Apr 26. Cell Death Differ. 2023. PMID: 37100955 Free PMC article. Review.
-
Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis.PLoS Genet. 2015 Mar 27;11(3):e1005092. doi: 10.1371/journal.pgen.1005092. eCollection 2015 Mar. PLoS Genet. 2015. PMID: 25816335 Free PMC article.
-
SIRT1-dependent myoprotective effects of resveratrol on muscle injury induced by compression.Front Physiol. 2015 Oct 21;6:293. doi: 10.3389/fphys.2015.00293. eCollection 2015. Front Physiol. 2015. PMID: 26557094 Free PMC article.
-
A Systematic Review of Oropharyngeal Dysphagia Models in Rodents.Int J Environ Res Public Health. 2021 May 7;18(9):4987. doi: 10.3390/ijerph18094987. Int J Environ Res Public Health. 2021. PMID: 34067192 Free PMC article.
References
-
- Abu-Baker A., Rouleau G.A. Oculopharyngeal muscular dystrophy: recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim. Biophys. Acta. 2007;1772:173–185. - PubMed
-
- Davies J.E., Berger Z., Rubinsztein D.C. Oculopharyngeal muscular dystrophy: potential therapies for an aggregate-associated disorder. Int. J. Biochem. Cell Biol. 2006;38:1457–1462. doi:10.1016/j.biocel.2006.01.016. - DOI - PubMed
-
- Schmitt H.P., Krause K.H. An autopsy study of a familial oculopharyngeal muscular dystrophy (OPMD) with distal spread and neurogenic involvement. Muscle Nerve. 1981;4:296–305. doi:10.1002/mus.880040406. - DOI - PubMed
-
- van der Sluijs B.M., van Engelen B.G., Hoefsloot L.H. Oculopharyngeal muscular dystrophy (OPMD) due to a small duplication in the PABPN1 gene. Hum. Mutat. 2003;21:553. doi:10.1002/humu.9138. - DOI - PubMed
-
- Blumen S.C., Brais B., Korczyn A.D., Medinsky S., Chapman J., Asherov A., Nisipeanu P., Codere F., Bouchard J.P., Fardeau M., et al. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann. Neurol. 1999;46:115–118. doi:10.1002/1531-8249(199907)46:1<115::AID-ANA17>3.0.CO;2-O. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
