

Amyloid-β inhibits No-cGMP signaling in a CD36- and CD47-dependent manner
- PMID: 21203512
- PMCID: PMC3008726
- DOI: 10.1371/journal.pone.0015686
Amyloid-β inhibits No-cGMP signaling in a CD36- and CD47-dependent manner
Abstract
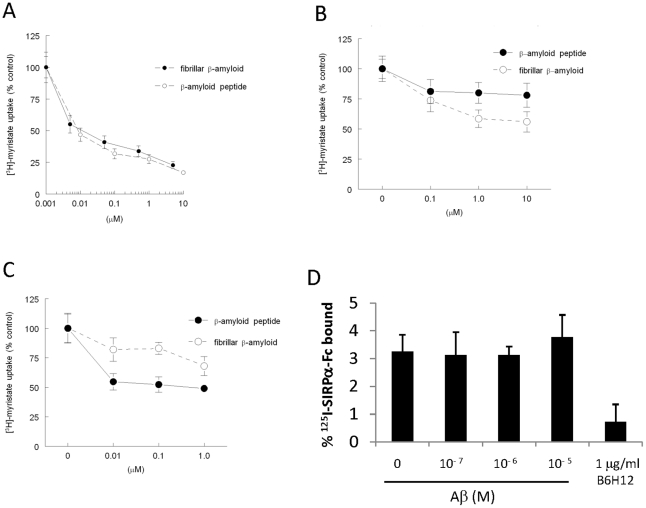
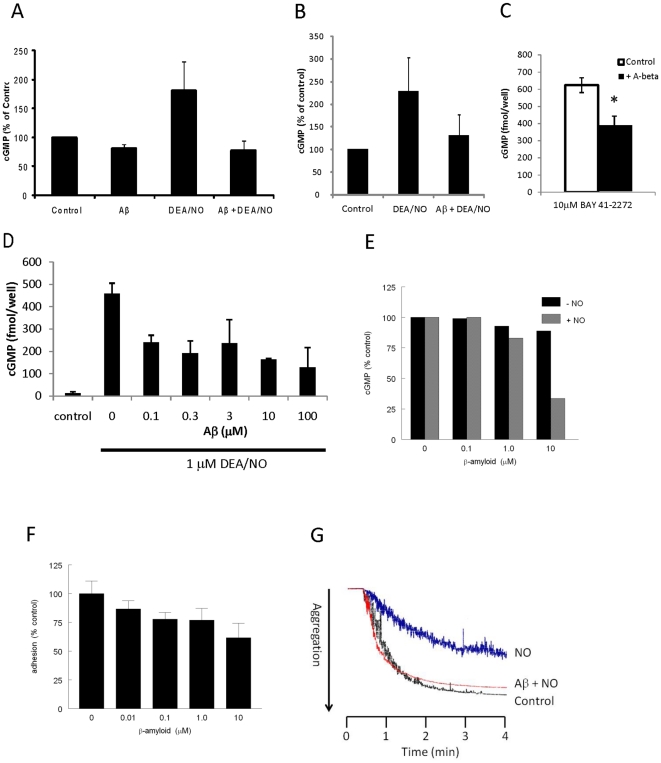
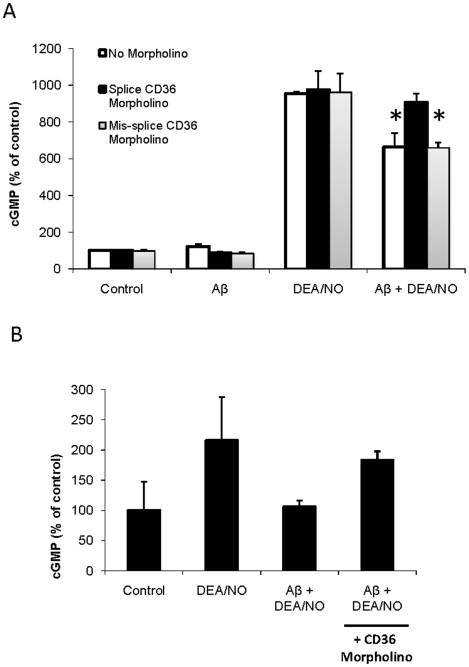
Amyloid-β interacts with two cell surface receptors, CD36 and CD47, through which the matricellular protein thrombospondin-1 inhibits soluble guanylate cyclase activation. Here we examine whether amyloid-β shares this inhibitory activity. Amyloid-β inhibited both drug and nitric oxide-mediated activation of soluble guanylate cyclase in several cell types. Known cGMP-dependent functional responses to nitric oxide in platelets and vascular smooth muscle cells were correspondingly inhibited by amyloid-β. Functional interaction of amyloid-β with the scavenger receptor CD36 was indicated by inhibition of free fatty acid uptake via this receptor. Both soluble oligomer and fibrillar forms of amyloid-β were active. In contrast, amyloid-β did not compete with the known ligand SIRPα for binding to CD47. However, both receptors were necessary for amyloid-β to inhibit cGMP accumulation. These data suggest that amyloid-β interaction with CD36 induces a CD47-dependent signal that inhibits soluble guanylate cyclase activation. Combined with the pleiotropic effects of inhibiting free fatty acid transport via CD36, these data provides a molecular mechanism through which amyloid-β can contribute to the nitric oxide signaling deficiencies associated with Alzheimer's disease.
Conflict of interest statement
Figures



References
-
- Selkoe DJ. Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci. 1994;17:489–517. - PubMed
-
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. - PubMed
-
- Verdier Y, Zarandi M, Penke B. Amyloid beta-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J Pept Sci. 2004;10:229–248. - PubMed
-
- Van Broeck B, Van Broeckhoven C, Kumar-Singh S. Current insights into molecular mechanisms of Alzheimer disease and their implications for therapeutic approaches. Neurodegener Dis. 2007;4:349–365. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
