

HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer
- PMID: 21203579
- PMCID: PMC3006345
- DOI: 10.1371/journal.pbio.1000563
HER2 phosphorylation is maintained by a PKB negative feedback loop in response to anti-HER2 herceptin in breast cancer
Erratum in
-
Correction: HER2 Phosphorylation Is Maintained by a PKB Negative Feedback Loop in Response to Anti-HER2 Herceptin in Breast Cancer.PLoS Biol. 2016 Mar 7;14(3):e1002414. doi: 10.1371/journal.pbio.1002414. eCollection 2016 Mar. PLoS Biol. 2016. PMID: 26949965 Free PMC article. No abstract available.
Abstract
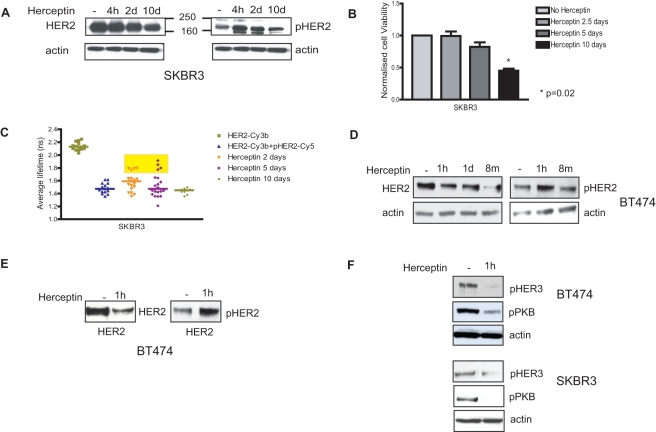
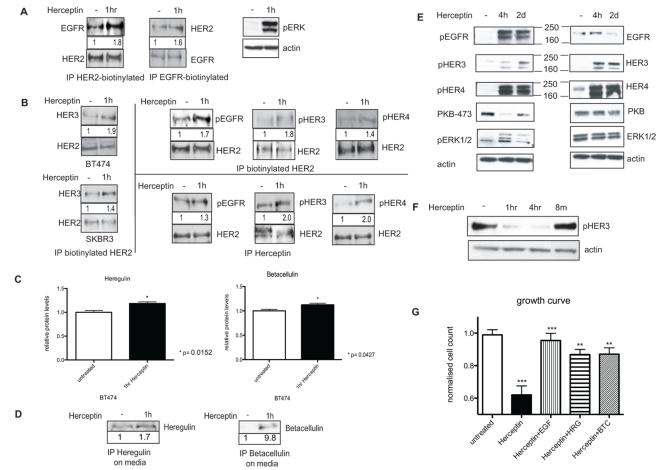
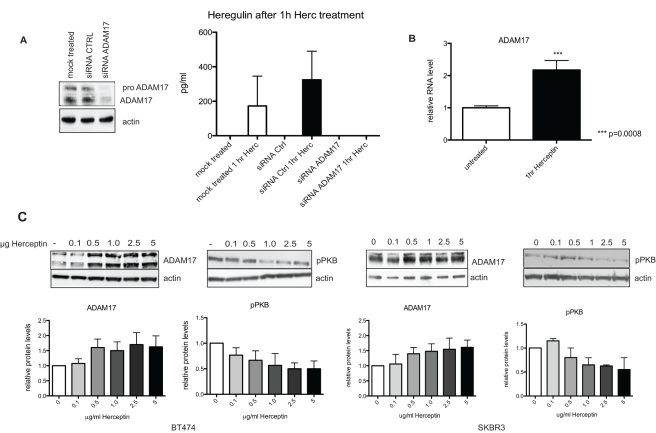
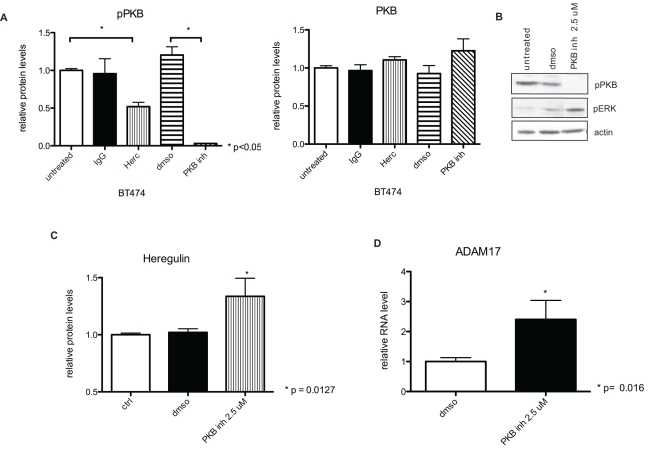
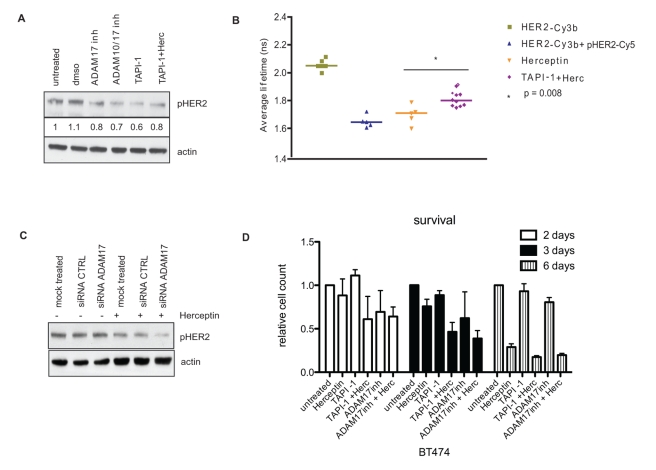
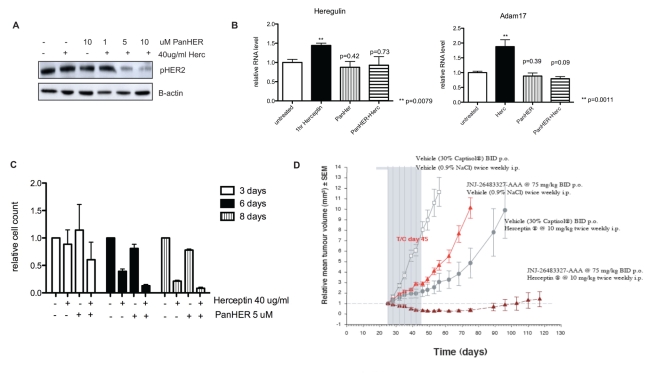
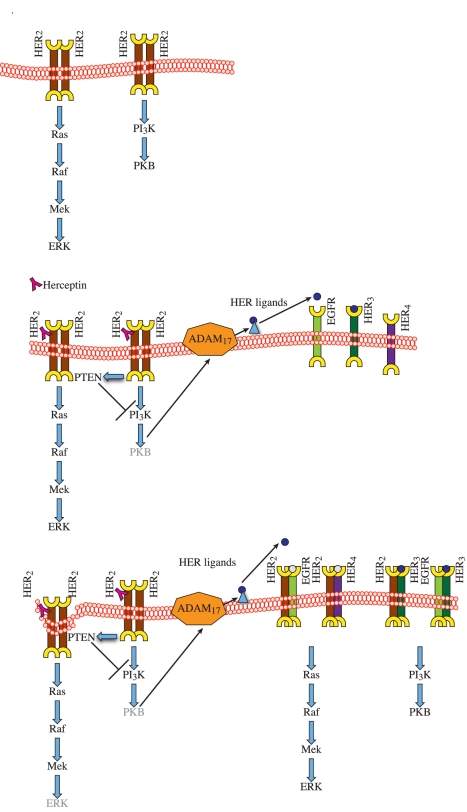
Herceptin (trastuzumab) is used in patients with breast cancer who have HER2 (ErbB2)-positive tumours. However, its mechanisms of action and how acquired resistance to Herceptin occurs are still poorly understood. It was previously thought that the anti-HER2 monoclonal antibody Herceptin inhibits HER2 signalling, but recent studies have shown that Herceptin does not decrease HER2 phosphorylation. Its failure to abolish HER2 phosphorylation may be a key to why acquired resistance inevitably occurs for all responders if Herceptin is given as monotherapy. To date, no studies have explained why Herceptin does not abolish HER2 phosphorylation. The objective of this study was to investigate why Herceptin did not decrease HER2 phosphorylation despite being an anti-HER2 monoclonal antibody. We also investigated the effects of acute and chronic Herceptin treatment on HER3 and PKB phosphorylation in HER2-positive breast cancer cells. Using both Förster resonance energy transfer (FRET) methodology and conventional Western blot, we have found the molecular mechanisms whereby Herceptin fails to abolish HER2 phosphorylation. HER2 phosphorylation is maintained by ligand-mediated activation of EGFR, HER3, and HER4 receptors, resulting in their dimerisation with HER2. The release of HER ligands was mediated by ADAM17 through a PKB negative feedback loop. The feedback loop was activated because of the inhibition of PKB by Herceptin treatment since up-regulation of HER ligands and ADAM17 also occurred when PKB phosphorylation was inhibited by a PKB inhibitor (Akt inhibitor VIII, Akti-1/2). The combination of Herceptin with ADAM17 inhibitors or the panHER inhibitor JNJ-26483327 was able to abrogate the feedback loop and decrease HER2 phosphorylation. Furthermore, the combination of Herceptin with JNJ-26483327 was synergistic in tumour inhibition in a BT474 xenograft model. We have determined that a PKB negative feedback loop links ADAM17 and HER ligands in maintaining HER2 phosphorylation during Herceptin treatment. The activation of other HER receptors via ADAM17 may mediate acquired resistance to Herceptin in HER2-overexpressing breast cancer. This finding offers treatment opportunities for overcoming resistance in these patients. We propose that Herceptin should be combined with a panHER inhibitor or an ADAM inhibitor to overcome the acquired drug resistance for patients with HER2-positive breast cancer. Our results may also have implications for resistance to other therapies targeting HER receptors.
Conflict of interest statement
Tim Perera and Peter King are employees and small share-holders of Johnson & Johnson Pharmaceutical Research & Development. This Competing Interest does not alter our adherence to all the PLoS Biology policies on sharing data and materials, or to the Creative Commons Attribution License in the event of publication.
Figures
References
-
- Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385. - PubMed
-
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. - PubMed
-
- Cho H. S, Mason K, Ramyar K. X, Stanley A. M, Gabelli S. B, et al. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature. 2003;421:756–760. - PubMed
-
- Slamon D. J, Clark G. M, Wong S. G, Levin W. J, Ullrich A, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
