

Fatty acid biosynthesis in actinomycetes
- PMID: 21204864
- PMCID: PMC3079561
- DOI: 10.1111/j.1574-6976.2010.00259.x
Fatty acid biosynthesis in actinomycetes
Abstract
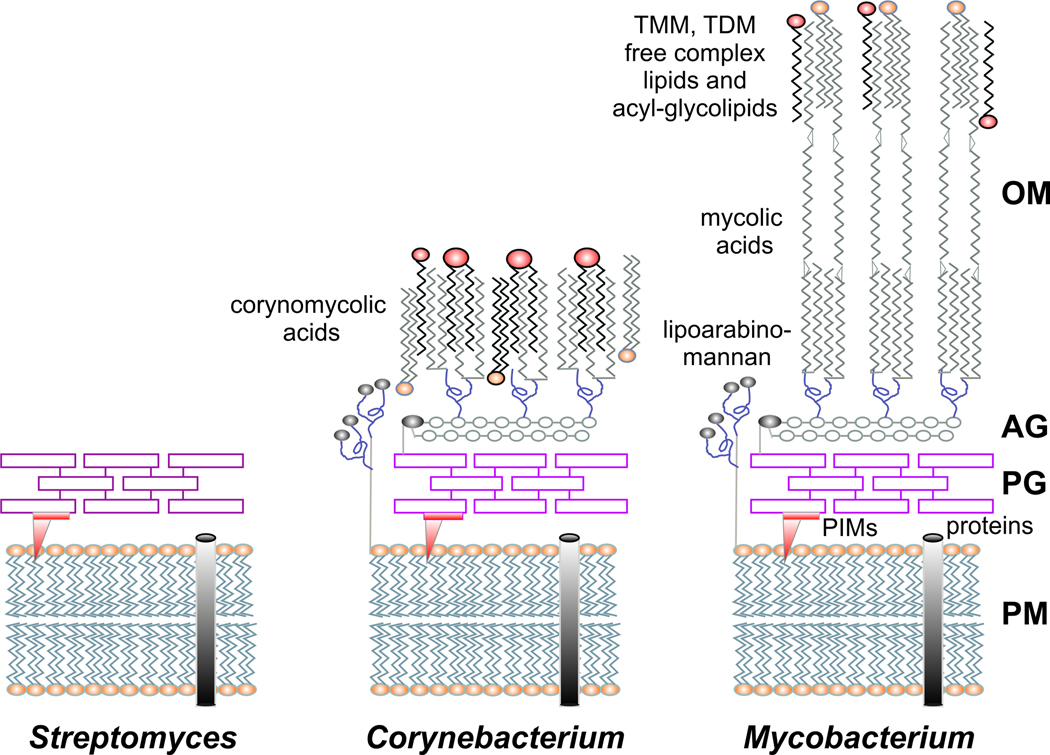
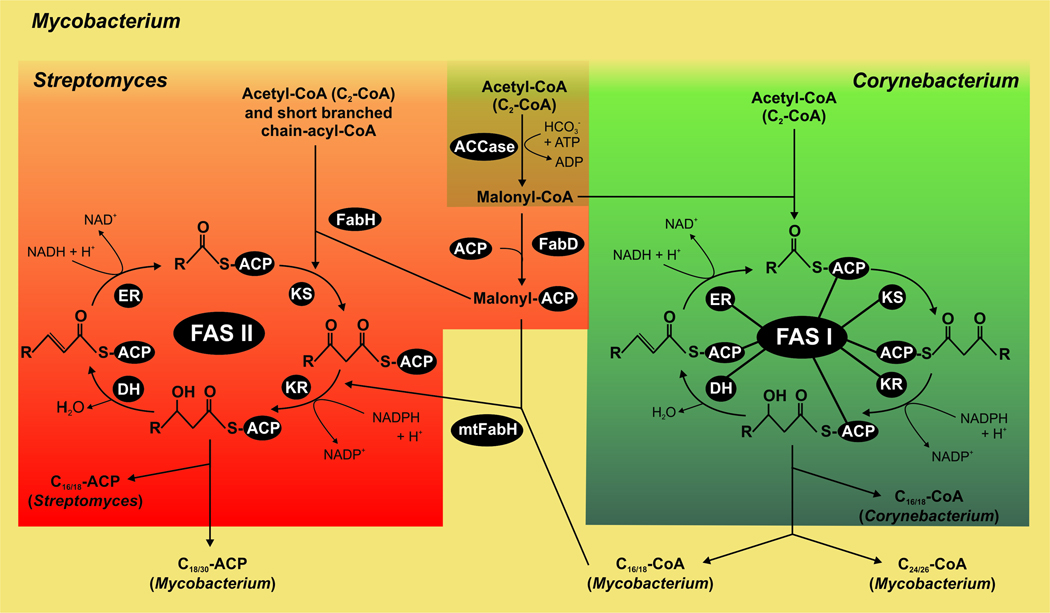
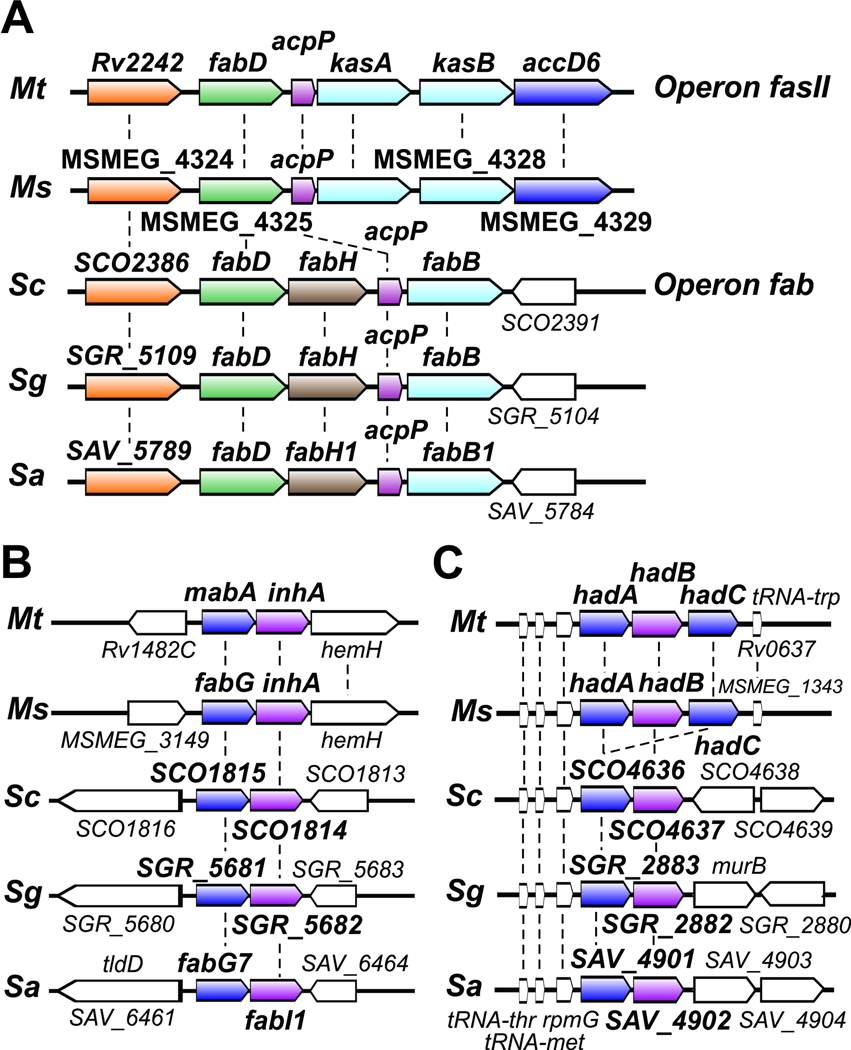
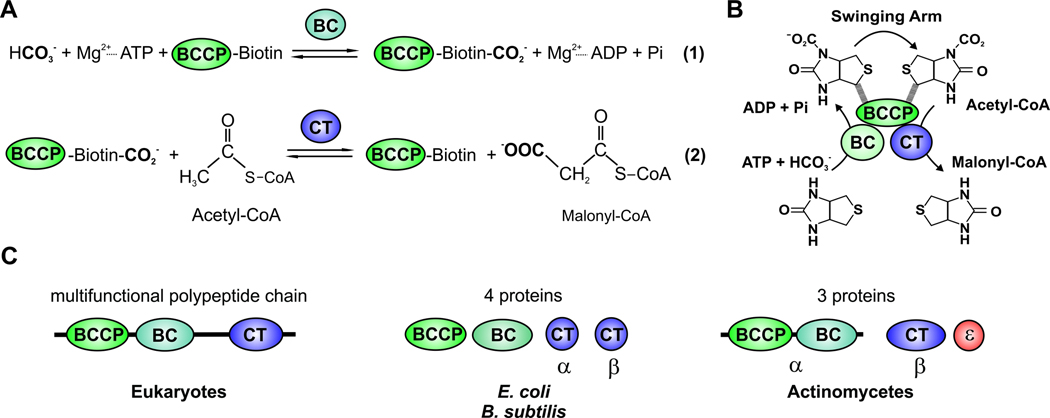
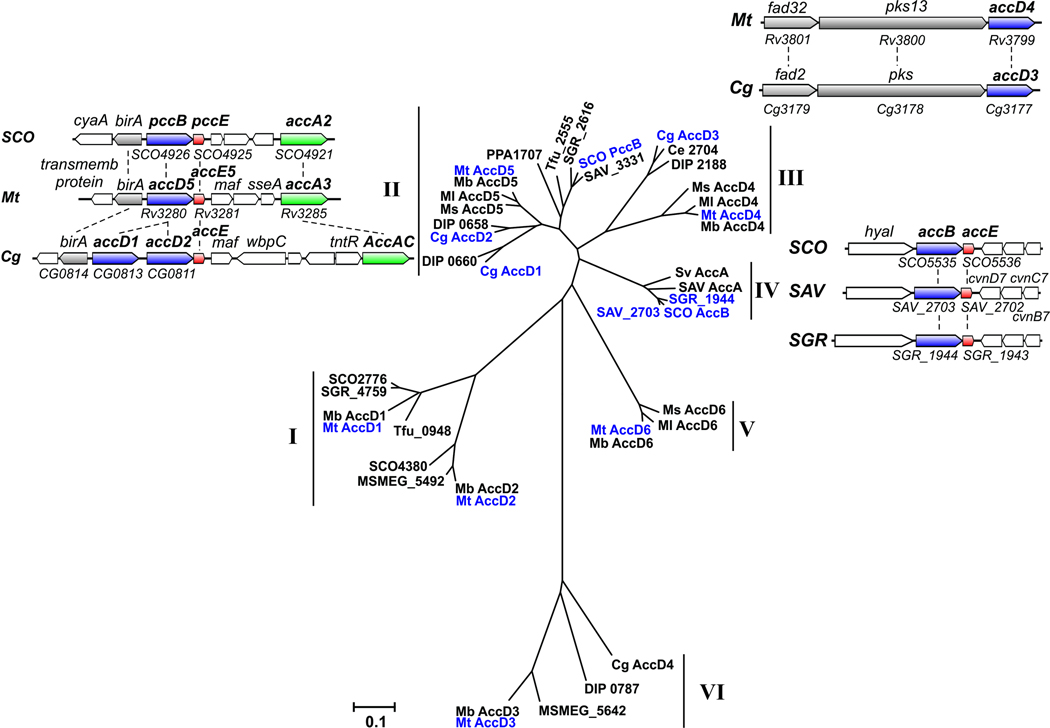
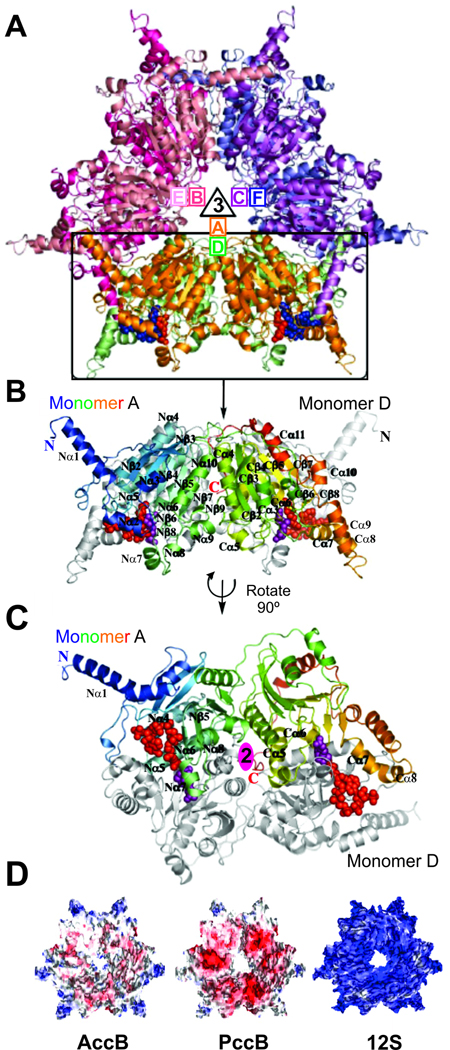
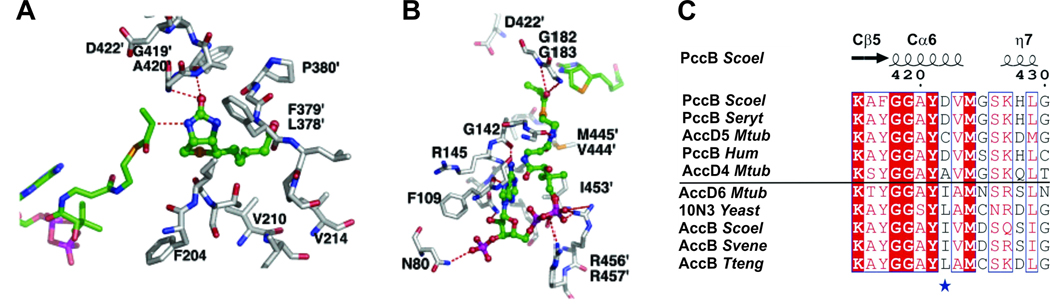
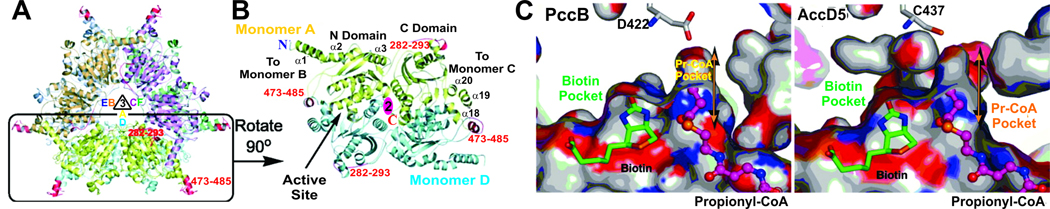
All organisms that produce fatty acids do so via a repeated cycle of reactions. In mammals and other animals, these reactions are catalyzed by a type I fatty acid synthase (FAS), a large multifunctional protein to which the growing chain is covalently attached. In contrast, most bacteria (and plants) contain a type II system in which each reaction is catalyzed by a discrete protein. The pathway of fatty acid biosynthesis in Escherichia coli is well established and has provided a foundation for elucidating the type II FAS pathways in other bacteria (White et al., 2005). However, fatty acid biosynthesis is more diverse in the phylum Actinobacteria: Mycobacterium, possess both FAS systems while Streptomyces species have only the multienzyme FAS II system and Corynebacterium species exclusively FAS I. In this review, we present an overview of the genome organization, biochemical properties and physiological relevance of the two FAS systems in the three genera of actinomycetes mentioned above. We also address in detail the biochemical and structural properties of the acyl-CoA carboxylases (ACCases) that catalyzes the first committed step of fatty acid synthesis in actinomycetes, and discuss the molecular bases of their substrate specificity and the structure-based identification of new ACCase inhibitors with antimycobacterial properties.
© 2011 Federation of European Microbiological Societies. Published by Blackwell Publishing Ltd. All rights reserved.
Figures
References
-
- Alberts AW, Bell RM, Vagelos PR. Acyl carrier protein. XV. Studies of - ketoacyl-acyl carrier protein synthetase. J Biol Chem. 1972;247:3190–3198. - PubMed
-
- Anderson AC. The process of structure-based drug design. Chem Biol. 2003;10:787–797. - PubMed
-
- Arabolaza A, D'Angelo M, Comba S, Gramajo H. FasR, a novel class of transcriptional regulator, governs the activation of fatty acid biosynthesis genes in Streptomyces coelicolor. Mol Microbiol. 2010a;78:47–63. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
