

Adaptive seeds tame genomic sequence comparison
- PMID: 21209072
- PMCID: PMC3044862
- DOI: 10.1101/gr.113985.110
Adaptive seeds tame genomic sequence comparison
Abstract
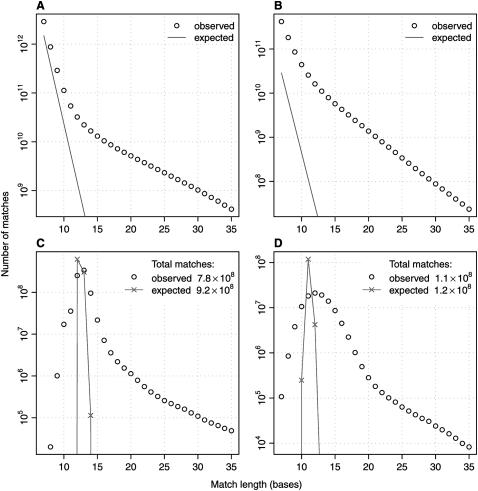
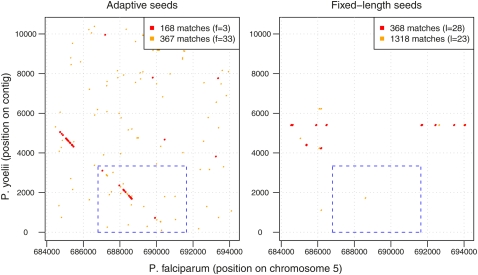
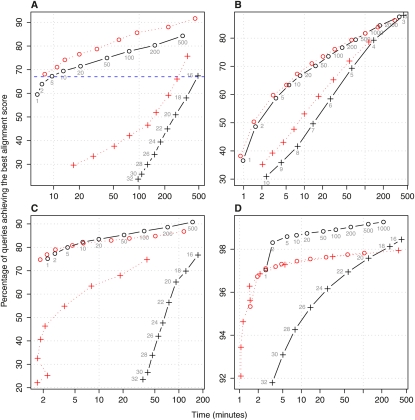
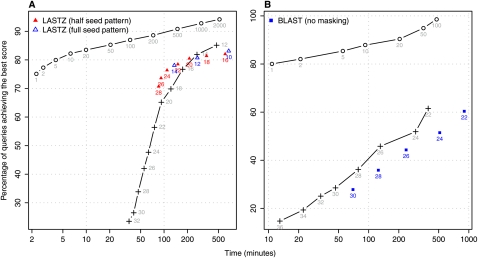
The main way of analyzing biological sequences is by comparing and aligning them to each other. It remains difficult, however, to compare modern multi-billionbase DNA data sets. The difficulty is caused by the nonuniform (oligo)nucleotide composition of these sequences, rather than their size per se. To solve this problem, we modified the standard seed-and-extend approach (e.g., BLAST) to use adaptive seeds. Adaptive seeds are matches that are chosen based on their rareness, instead of using fixed-length matches. This method guarantees that the number of matches, and thus the running time, increases linearly, instead of quadratically, with sequence length. LAST, our open source implementation of adaptive seeds, enables fast and sensitive comparison of large sequences with arbitrarily nonuniform composition.
Figures
References
-
- Abouelhoda MI, Kurtz S, Ohlebusch E 2004. Replacing suffix trees with enhanced suffix arrays. J Discrete Algorithms 2: 53–86
-
- Batzer MA, Deininger PL 2002. Alu repeats and human genomic diversity. Nat Rev Genet 3: 370–379 - PubMed
-
- Carlton J, Silva J, Hall N 2005. The genome of model malaria parasites, and comparative genomics. Curr Issues Mol Biol 7: 23–37 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials