

Proteasome activators
- PMID: 21211719
- PMCID: PMC3040445
- DOI: 10.1016/j.molcel.2010.12.020
Proteasome activators
Abstract
Proteasomes degrade a multitude of protein substrates in the cytosol and nucleus, and thereby are essential for many aspects of cellular function. Because the proteolytic sites are sequestered in a closed barrel-shaped structure, activators are required to facilitate substrate access. Structural and biochemical studies of two activator families, 11S and Blm10, have provided insights to proteasome activation mechanisms, although the biological functions of these factors remain obscure. Recent advances have improved our understanding of the third activator family, including the 19S activator, which targets polyubiquitylated proteins for degradation. Here we present a structural perspective on how proteasomes are activated and how substrates are delivered to the proteolytic sites.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
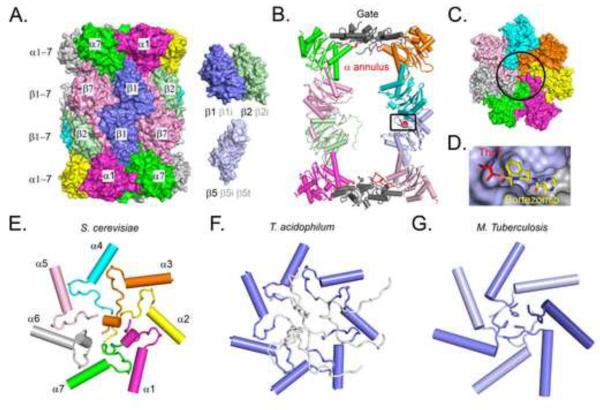
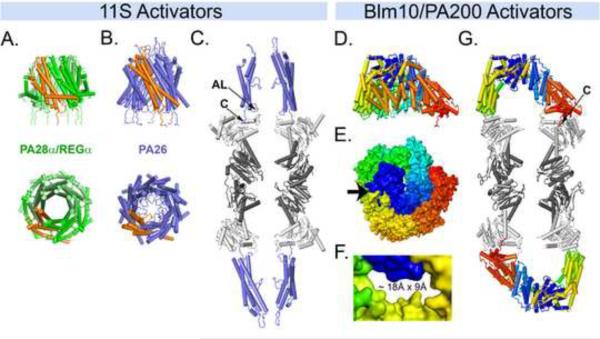
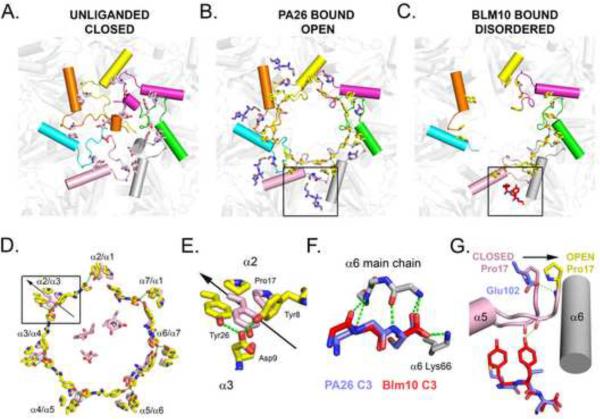
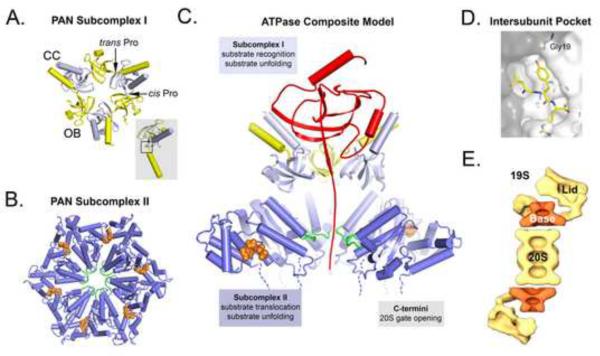
References
-
- Babbitt SE, Kiss A, Deffenbaugh AE, Chang YH, Bailly E, Erdjument-Bromage H, Tempst P, Buranda T, Sklar LA, Baumler J, et al. ATP hydrolysis-dependent disassembly of the 26S proteasome is part of the catalytic cycle. Cell. 2005;121:553–565. - PubMed
-
- Bajorek M, Finley D, Glickman MH. Proteasome disassembly and downregulation is correlated with viability during stationary phase. Curr Biol. 2003;13:1140–1144. - PubMed
-
- Bech-Otschir D, Helfrich A, Enenkel C, Consiglieri G, Seeger M, Holzhutter HG, Dahlmann B, Kloetzel PM. Polyubiquitin substrates allosterically activate their own degradation by the 26S proteasome. Nat Struct Mol Biol. 2009;16:219–225. - PubMed
-
- Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell. 2003;11:69–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
