

GABA and central neuropathic pain following spinal cord injury
- PMID: 21216257
- PMCID: PMC3285561
- DOI: 10.1016/j.neuropharm.2010.12.030
GABA and central neuropathic pain following spinal cord injury
Abstract
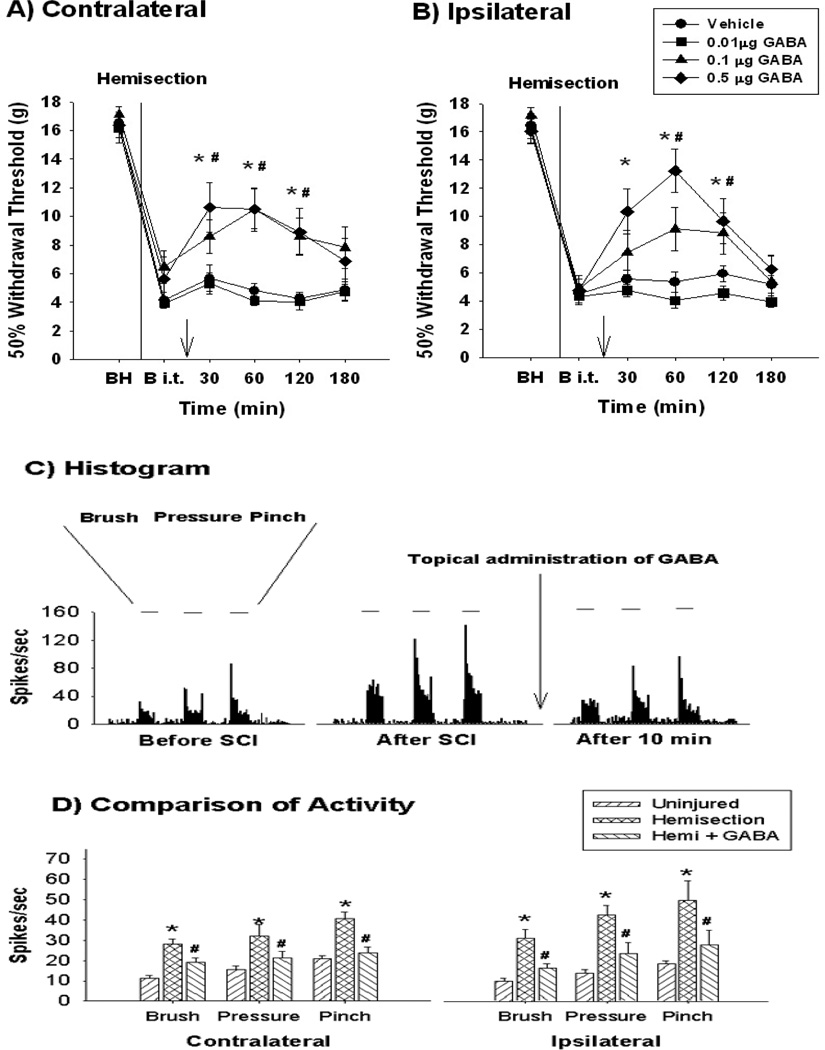
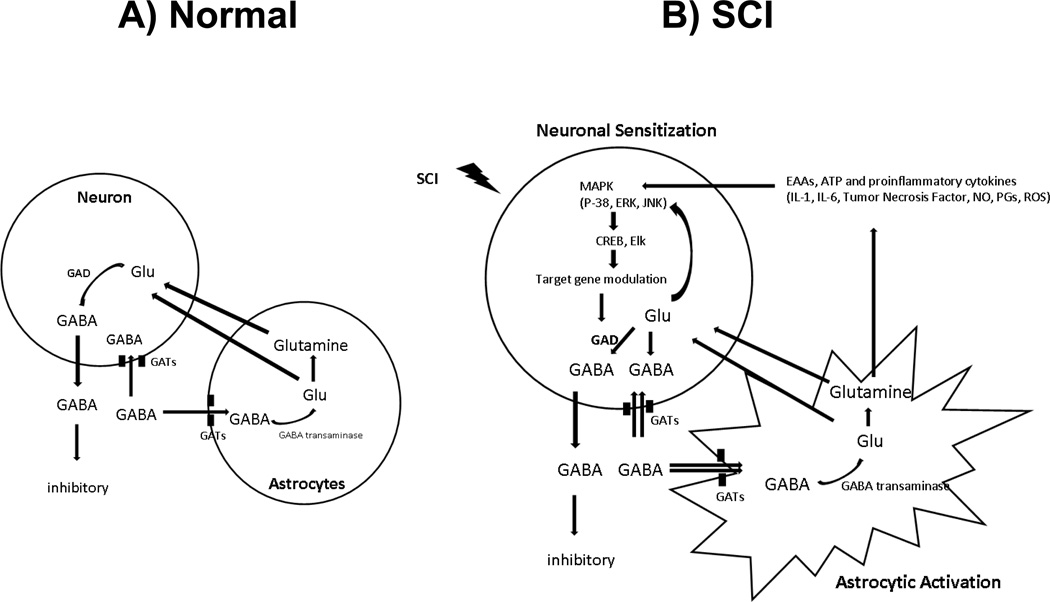
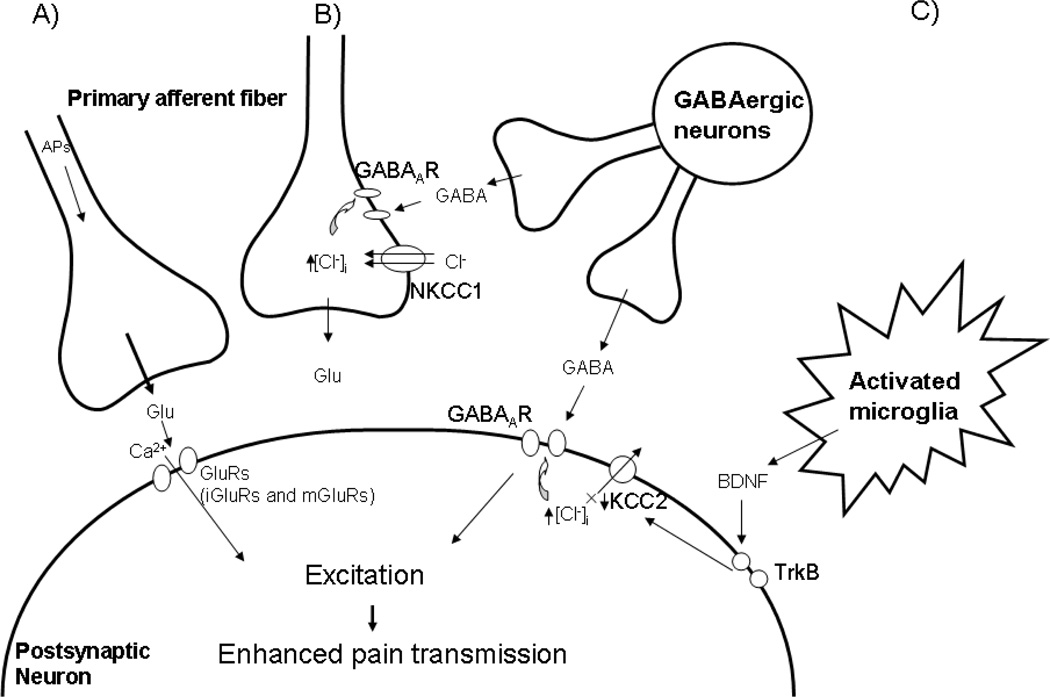
Spinal cord injury induces maladaptive synaptic transmission in the somatosensory system that results in chronic central neuropathic pain. Recent literature suggests that glial-neuronal interactions are important modulators in synaptic transmission following spinal cord injury. Neuronal hyperexcitability is one of the predominant phenomenon caused by maladaptive synaptic transmission via altered glial-neuronal interactions after spinal cord injury. In the somatosensory system, spinal inhibitory neurons counter balance the enhanced synaptic transmission from peripheral input. For a decade, the literature suggests that hypofunction of GABAergic inhibitory tone is an important factor in the enhanced synaptic transmission that often results in neuronal hyperexcitability in dorsal horn neurons following spinal cord injury. Neurons and glial cells synergistically control intracellular chloride ion gradients via modulation of chloride transporters, extracellular glutamate and GABA concentrations via uptake mechanisms. Thus, the intracellular "GABA-glutamate-glutamine cycle" is maintained for normal physiological homeostasis. However, hyperexcitable neurons and glial activation after spinal cord injury disrupts the balance of chloride ions, glutamate and GABA distribution in the spinal dorsal horn and results in chronic neuropathic pain. In this review, we address spinal cord injury induced mechanisms in hypofunction of GABAergic tone that results in chronic central neuropathic pain. This article is part of a Special Issue entitled 'Synaptic Plasticity & Interneurons'.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Disclosure
No conflict of interest
Figures
References
-
- Albrecht J, Pearce B, Murphy S. Evidence for an interaction between GABAB and glutamate receptors in astrocytes as revealed by changes in Ca2+ flux. Eur. J. Pharmacol. 1986;125:463–464. - PubMed
-
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. - PubMed
-
- Aptel H, Hilaire C, Pieraut S, Boukhaddaoui H, Mallie S, Valmier J, Scamps F. The Cav3.2/alpha1H T-type Ca2+ current is a molecular determinant of excitatory effects of GABA in adult sensory neurons. Mol. Cell. Neurosci. 2007;36:293–303. - PubMed
-
- Bao F, Liu D. Peroxynitrite generated in the rat spinal cord induces apoptotic cell death and activates caspase-3. Neuroscience. 2003;116:59–70. - PubMed
-
- Barakat L, Bordey A. GAT-1 and reversible GABA transport in Bergmann glia in slices. J. Neurophysiol. 2002;88:1407–1419. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
