

Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome
- PMID: 21220633
- PMCID: PMC3952880
- DOI: 10.1001/archophthalmol.2010.330
Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome
Abstract
Objective: To investigate whether mutations in NPHP5 can cause Leber congenital amaurosis (LCA) without early-onset renal disease.
Methods: DNA samples from 276 individuals with nonsyndromic LCA were screened for variations in the NPHP5 gene. Each had been previously screened for mutations in 8 known LCA genes without identifying a disease-causing genotype.
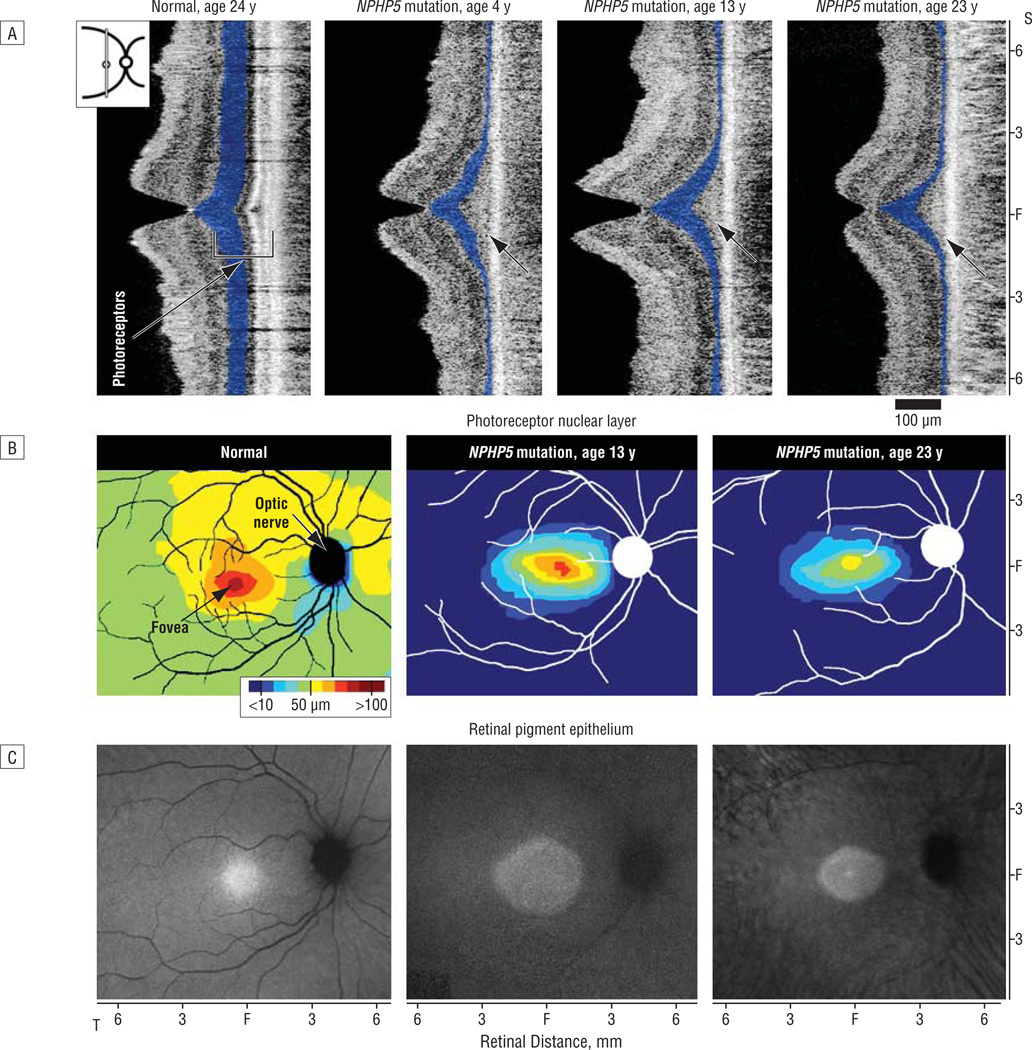
Results: Nine of the 276 LCA probands (3.2%) harbored 2 plausible disease-causing mutations (7 different alleles) in NPHP5. Four of these have been previously reported in patients with Senior-Loken syndrome (F141del, R461X, H506del, and R489X) and 3 are novel (A111del, E346X, and R455X). All 9 patients had severe visual loss from early childhood but none had overt renal disease in the first decade of life. Two patients were diagnosed with nephronophthisis in the second decade. Retinal imaging studies showed retained photoreceptor nuclei and retinal pigment epithelium integrity mainly in the cone-rich central retina, a phenotype with strong similarities to that of NPHP6 disease.
Conclusions: Mutations in NPHP5 can cause LCA without early-onset renal disease. Abnormalities observed in the photoreceptor outer segments (a cilial structure) may explain the severe visual loss in NPHP5 -associated LCA. Clinical Relevance The persistence of central photoreceptor nuclei despite severe visual loss in NPHP5 disease is encouraging for future therapeutic interventions.
Figures

References
-
- Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139(4):663–678. - PubMed
-
- Cardenas-Rodriguez M, Badano JL. Ciliary biology: understanding the cellular and genetic basis of human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C(4):263–280. - PubMed
-
- Senior B, Friedmann AI, Braudo JL. Juvenile familial nephropathy with tapeto-retinal degeneration. Am J Ophthalmol. 1961;52:625–633. - PubMed
-
- Loken AC, Hanssen O, Halvorsen S, Jolster NJ. Hereditary renal dysplasia and blindness. Acta Paediatr. 1961;50:177–184. - PubMed
-
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18(6):1855–1871. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
