

AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision
- PMID: 21233167
- PMCID: PMC3042181
- DOI: 10.1093/bioinformatics/btq713
AGE: defining breakpoints of genomic structural variants at single-nucleotide resolution, through optimal alignments with gap excision
Abstract
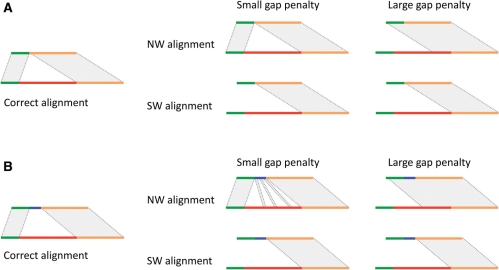
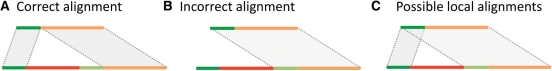
Motivation: Defining the precise location of structural variations (SVs) at single-nucleotide breakpoint resolution is an important problem, as it is a prerequisite for classifying SVs, evaluating their functional impact and reconstructing personal genome sequences. Given approximate breakpoint locations and a bridging assembly or split read, the problem essentially reduces to finding a correct sequence alignment. Classical algorithms for alignment and their generalizations guarantee finding the optimal (in terms of scoring) global or local alignment of two sequences. However, they cannot generally be applied to finding the biologically correct alignment of genomic sequences containing SVs because of the need to simultaneously span the SV (e.g. make a large gap) and perform precise local alignments at the flanking ends.
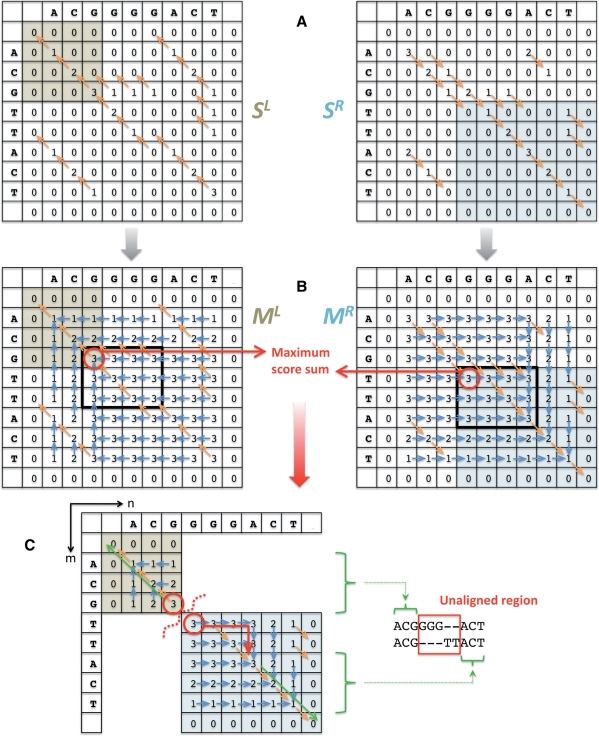
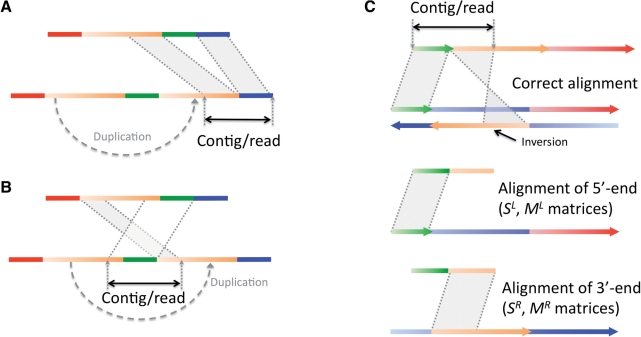
Results: Here, we formulate the computations involved in this problem and describe a dynamic-programming algorithm for its solution. Specifically, our algorithm, called AGE for Alignment with Gap Excision, finds the optimal solution by simultaneously aligning the 5' and 3' ends of two given sequences and introducing a 'large-gap jump' between the local end alignments to maximize the total alignment score. We also describe extensions allowing the application of AGE to tandem duplications, inversions and complex events involving two large gaps. We develop a memory-efficient implementation of AGE (allowing application to long contigs) and make it available as a downloadable software package. Finally, we applied AGE for breakpoint determination and standardization in the 1000 Genomes Project by aligning locally assembled contigs to the human genome.
Availability and implementation: AGE is freely available at http://sv.gersteinlab.org/age.
Figures

References
-
- Chao KM, et al. Recent developments in linear-space alignment methods: a survey. J. Comput. Biol. 1994;1:271–291. - PubMed
-
- Gotoh O. An improved algorithm for matching biological sequences. J. Mol. Biol. 1982;162:705–708. - PubMed
-
- Hirschberg DS. A linear space algorithm for computing maximal common subsequences. Commun. ACM. 1975;18:341–343.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
