

Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy
- PMID: 21233847
- PMCID: PMC3131943
- DOI: 10.1038/cdd.2010.177
Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy
Abstract
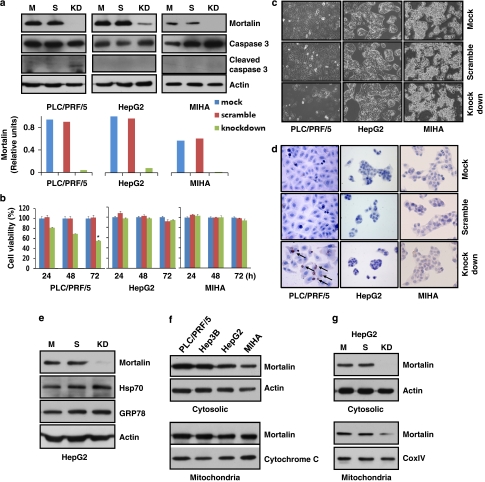
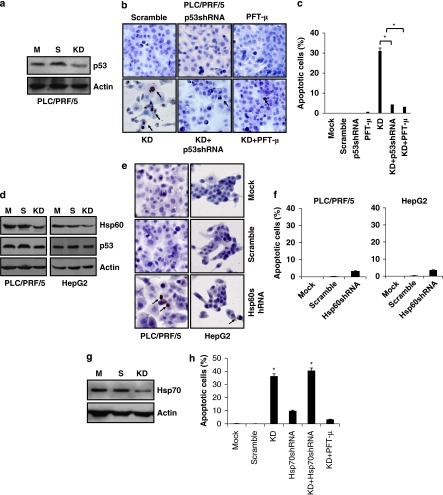
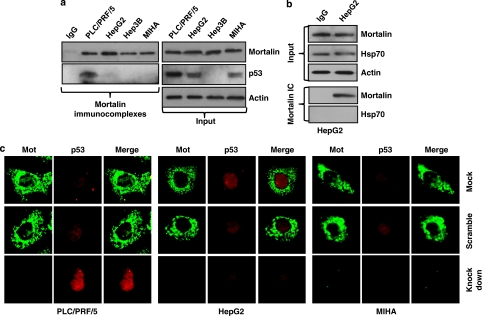
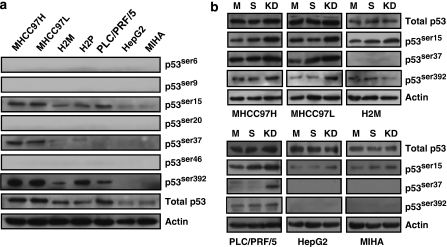
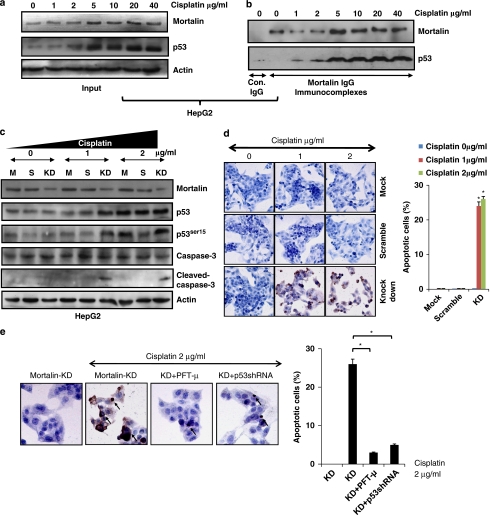
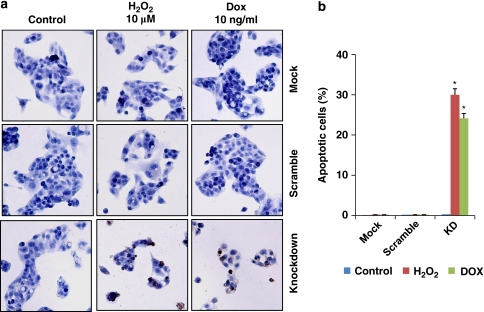
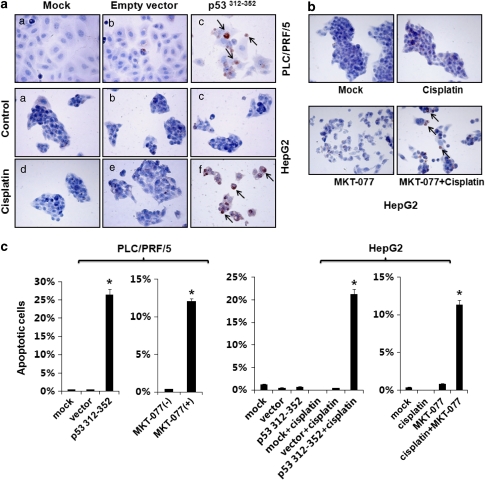
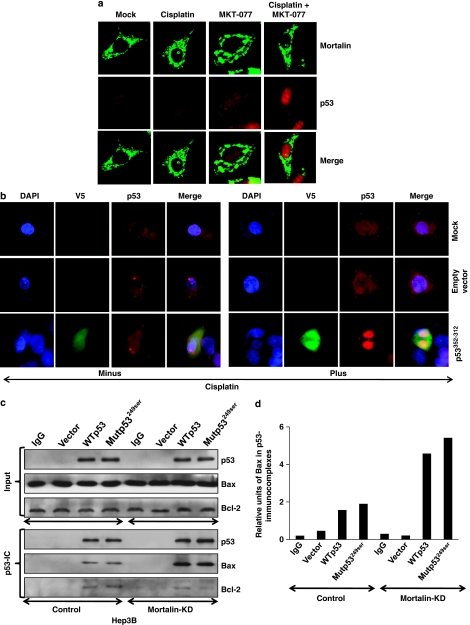
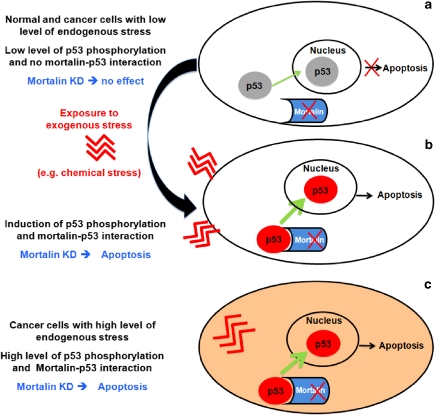
Stress protein mortalin is a multifunctional protein and is highly expressed in cancers. It has been shown to interact with tumor suppressor protein-p53 (both wild and mutant types) and inactivates its transcriptional activation and apoptotic functions in cancer cells. In the present study, we found that, unlike most of the cancer cells, HepG2 hepatoma lacked mortalin-p53 interaction. We demonstrate that the mortalin-p53 interaction exists in cancer cells that are either physiologically stressed (frequently associated with p53 mutations) or treated with stress-inducing chemicals. Targeting mortalin-p53 interaction with either mortalin small hairpin RNA or a chemical or peptide inhibitor could induce p53-mediated tumor cell-specific apoptosis in hepatocellular carcinoma; p53-null hepatoma or normal hepatocytes remain unaffected.
Figures
References
-
- Kastan MB, Zhan QM, Eldeiry WS, Carrier F, Jacks T, Walsh WV, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. - PubMed
-
- Yonish-Rouach E, Resnitzky D, Lotem J, Sachs L, Kimchi A, Oren M. Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature. 1991;352:345–347. - PubMed
-
- Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell. 2003;112:29–40. - PubMed
-
- Liu S, Li J, Tao Y, Xiao X. Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem Biophys Res Commun. 2007;354:109–114. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
