

Apoptosis and oncogenesis: give and take in the BCL-2 family
- PMID: 21236661
- PMCID: PMC3040981
- DOI: 10.1016/j.gde.2010.12.001
Apoptosis and oncogenesis: give and take in the BCL-2 family
Abstract
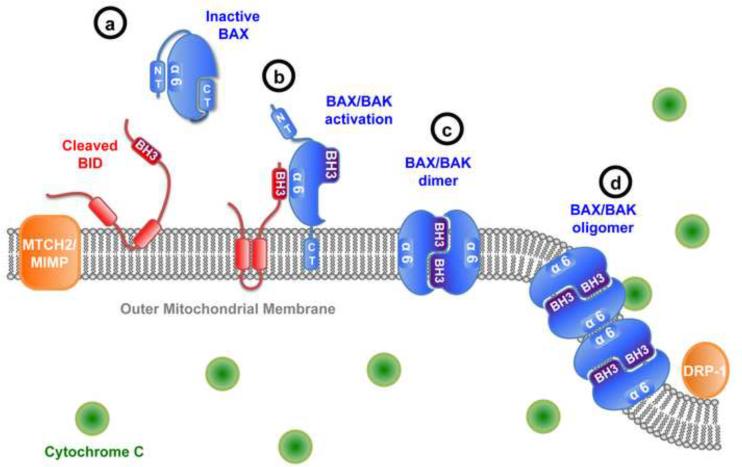
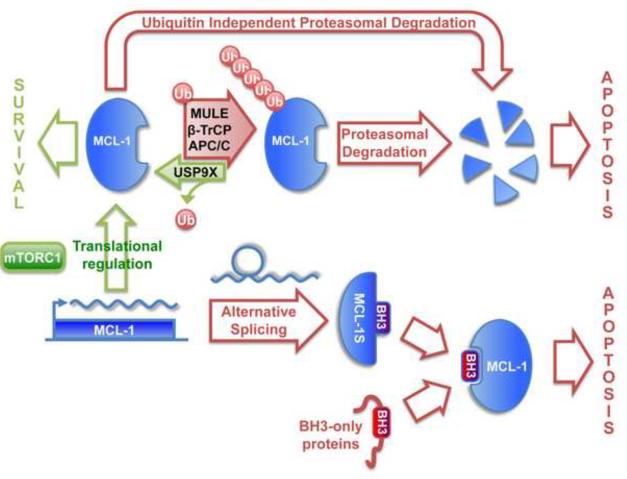
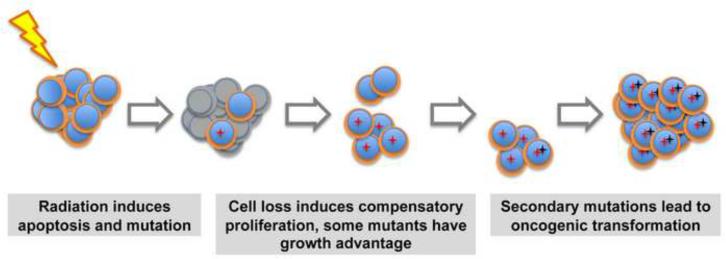
The mitochondrial pathway of apoptosis constitutes one of the main safeguards against tumorigenesis. The BCL-2 family includes the central players of this pathway that regulate cell fate through the control of mitochondrial outer membrane permeabilization (MOMP), and important progress has been made in understanding the dynamic interactions between pro-apoptotic and anti-apoptotic BCL-2 proteins. In particular, recent studies have delineated a stepwise model for the induction of MOMP. BCL-2 proteins are often dysregulated in cancer, leading to increased survival of abnormal cells; however, recent studies have paradoxically shown that apoptosis induction can under some circumstances drive tumor formation, perhaps by inducing compensatory proliferation under conditions of cellular stress. These observations underline the complexity of BCL-2 protein function in oncogenesis.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
