

Nicotine aggravates the brain postischemic inflammatory response
- PMID: 21239632
- PMCID: PMC3075028
- DOI: 10.1152/ajpheart.00928.2010
Nicotine aggravates the brain postischemic inflammatory response
Abstract
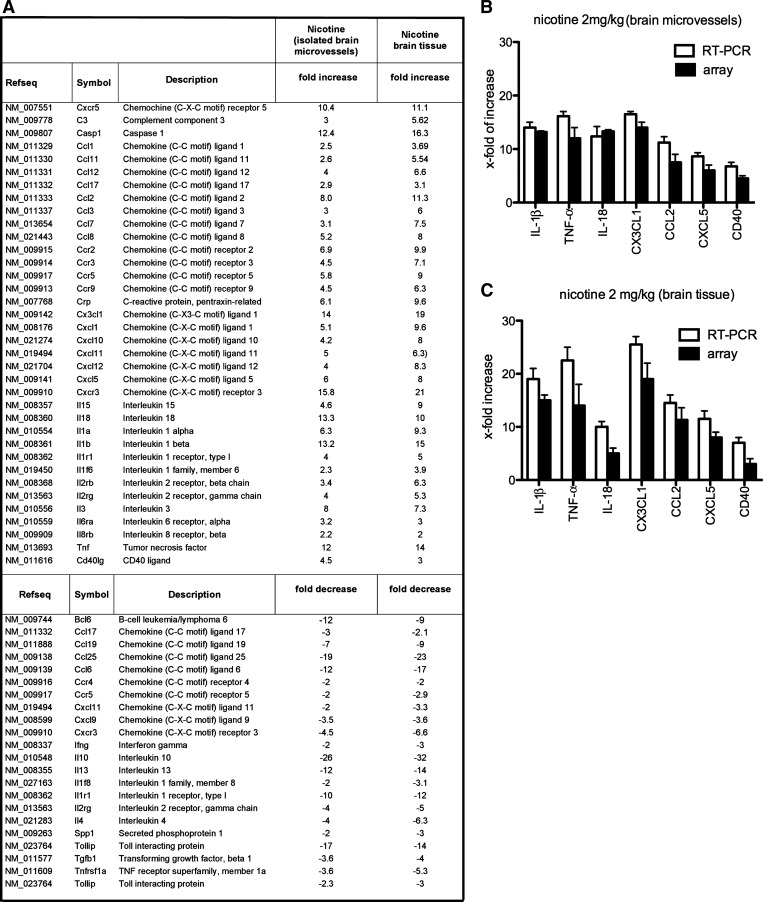
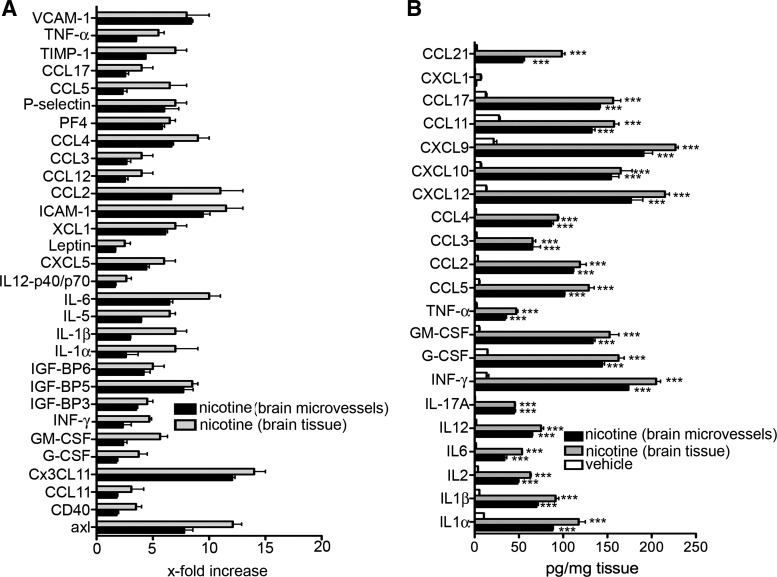
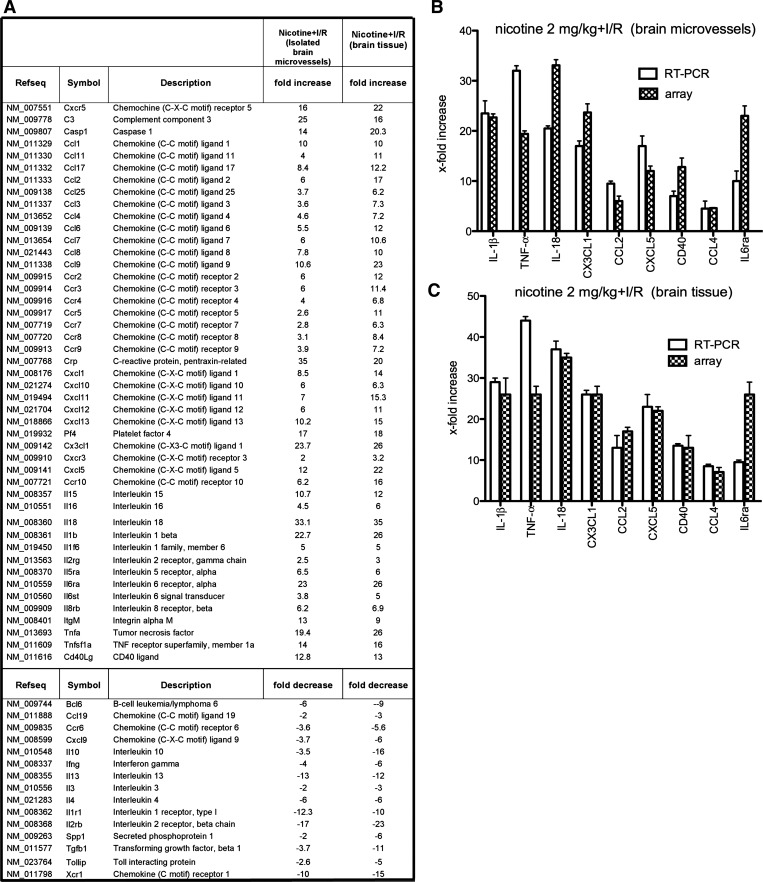
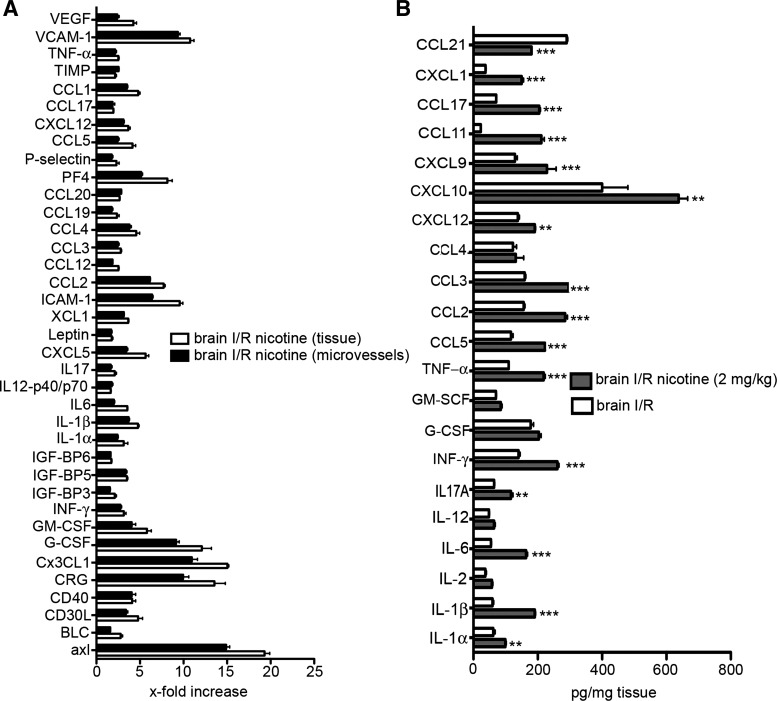
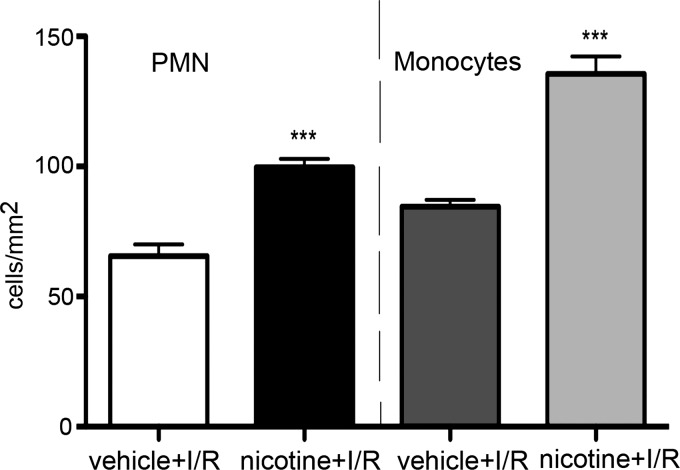
A substantial body of evidence suggests that nicotine adversely affects cerebral blood flow and the blood-brain barrier and is a risk factor for stroke. The present study investigated the effect of nicotine on cerebrovascular endothelium under basal and ischemia/reperfusion injury under in vivo condition. Nicotine (2 mg/kg sc) was administered to mice over 14 days, which resulted in plasma nicotine levels of ∼100 ng/ml, reflecting plasma concentrations in average to heavy smokers. An analysis of the phenotype of isolated brain microvessels after nicotine exposure indicated higher expression of inflammatory mediators, cytokines (IL-1β, TNF-α, and IL-18), chemokines (CCL2 and CX(3)CL1), and adhesion molecules (ICAM-1, VCAM-1, and P-selectins), and this was accompanied by enhanced leukocyte infiltration into brain during ischemia/reperfusion (P < 0.01). Nicotine had a profound effect on ischemia/reperfusion injury; i.e., increased brain infarct size (P < 0.01), worse neurological deficits, and a higher mortality rate. These experiments illuminate, for the first time, how nicotine regulates brain endothelial cell phenotype and postischemic inflammatory response at the brain-vascular interface.
Figures

References
-
- Abbruscato TJ, Lopez SP, Roder K, Paulson JR. Regulation of blood-brain barrier Na,K,2Cl-cotransporter through phosphorylation during in vitro stroke conditions and nicotine exposure. J Pharmacol Exp Ther 310: 459– 468, 2004. - PubMed
-
- Albaugh G, Bellavance E, Strande L, Heinburger S, Hewitt CW, Alexander JB. Nicotine induces mononuclear leukocyte adhesion and expression of adhesion molecules, VCAM and ICAM, in endothelial cells in vitro. Ann Vasc Surg 18: 302– 307, 2004. - PubMed
-
- Albaugh G, Kann B, Strande L, Vemulapalli P, Hewitt C, Alexander JB. Nicotine induces endothelial TNF-alpha expression, which mediates growth retardation in vitro. J Surg Res 99: 381– 384, 2001. - PubMed
-
- Andjelkovic AV, Stamatovic SM, Keep RF. The protective effects of preconditioning on cerebral endothelial cells in vitro. J Cereb Blood Flow Metab 23: 1348– 1355, 2003. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous