

Densely interconnected transcriptional circuits control cell states in human hematopoiesis
- PMID: 21241896
- PMCID: PMC3049864
- DOI: 10.1016/j.cell.2011.01.004
Densely interconnected transcriptional circuits control cell states in human hematopoiesis
Abstract
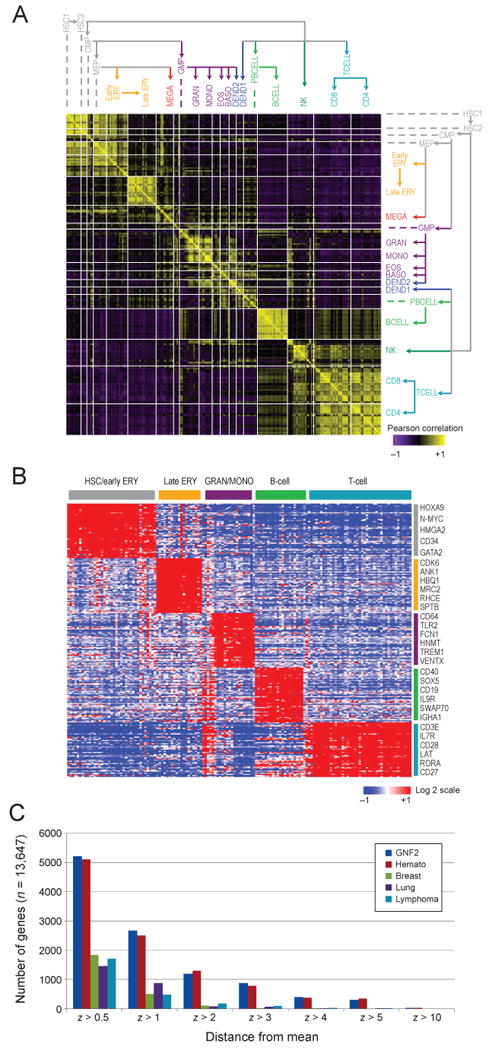
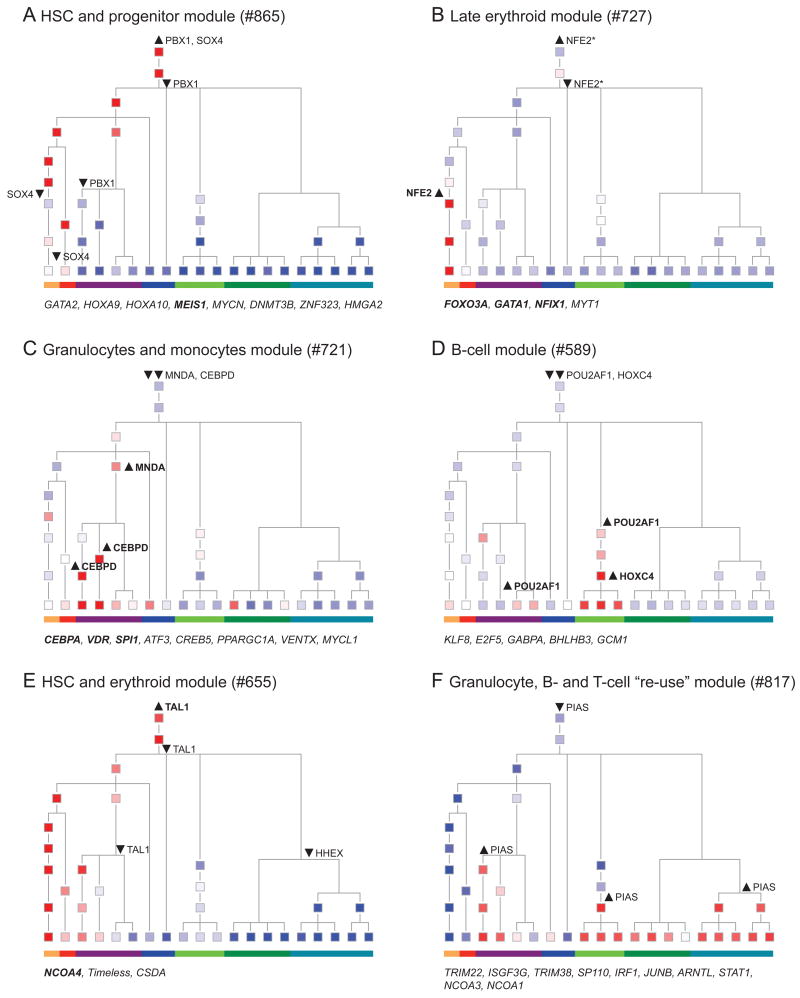
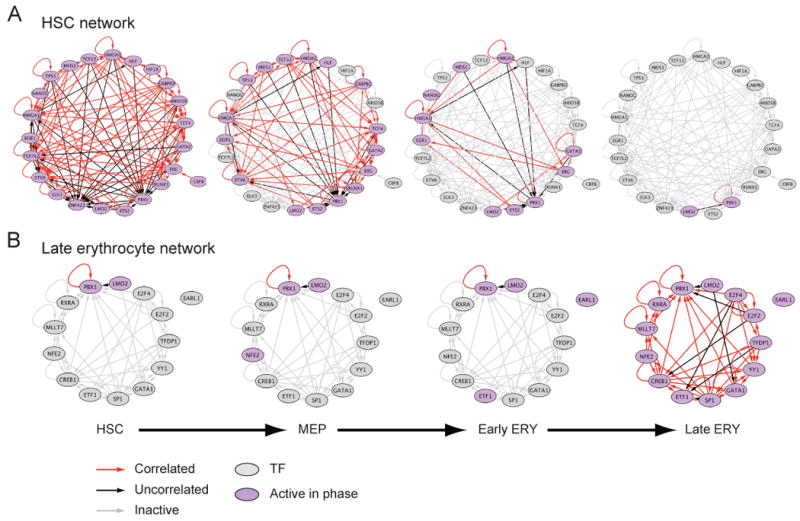
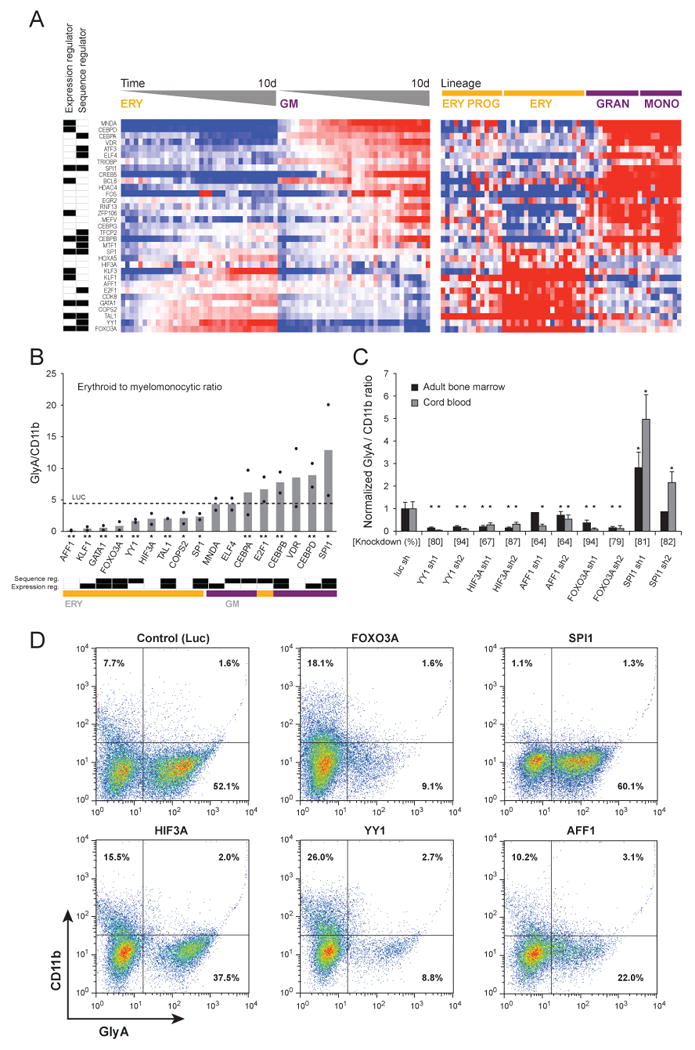
Though many individual transcription factors are known to regulate hematopoietic differentiation, major aspects of the global architecture of hematopoiesis remain unknown. Here, we profiled gene expression in 38 distinct purified populations of human hematopoietic cells and used probabilistic models of gene expression and analysis of cis-elements in gene promoters to decipher the general organization of their regulatory circuitry. We identified modules of highly coexpressed genes, some of which are restricted to a single lineage but most of which are expressed at variable levels across multiple lineages. We found densely interconnected cis-regulatory circuits and a large number of transcription factors that are differentially expressed across hematopoietic states. These findings suggest a more complex regulatory system for hematopoiesis than previously assumed.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures



Comment in
-
A comprehensive transcriptional landscape of human hematopoiesis.Cell Stem Cell. 2011 Feb 4;8(2):122-4. doi: 10.1016/j.stem.2011.01.006. Cell Stem Cell. 2011. PMID: 21295265
References
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
