

Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis
- PMID: 21242297
- PMCID: PMC3039852
- DOI: 10.1084/jem.20100370
Growth differentiation factor 15 deficiency protects against atherosclerosis by attenuating CCR2-mediated macrophage chemotaxis
Abstract
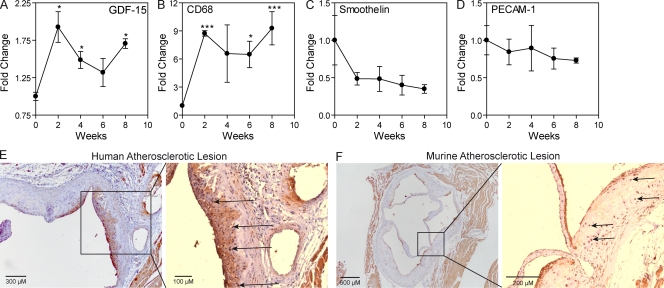
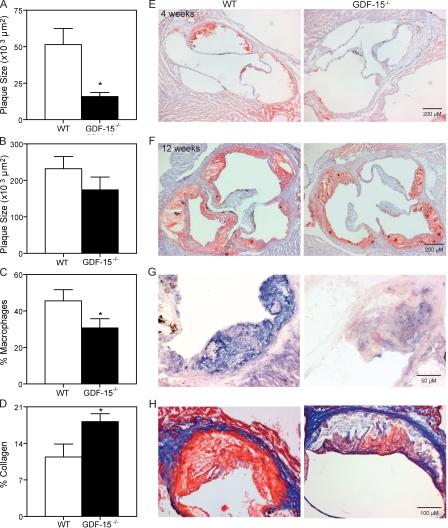
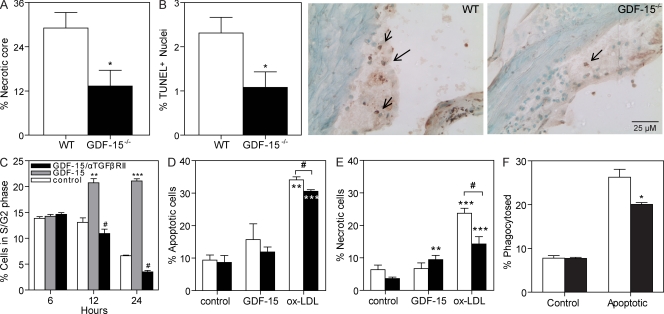
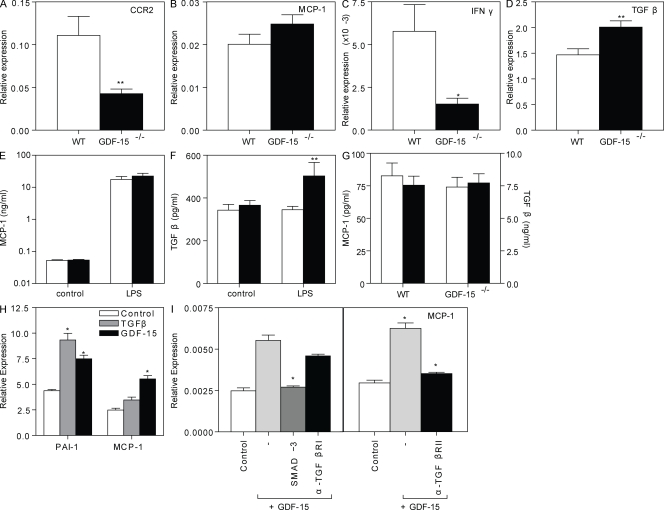
Growth differentiation factor (GDF) 15 is a member of the transforming growth factor β (TGF-β) superfamily, which operates in acute phase responses through a currently unknown receptor. Elevated GDF-15 serum levels were recently identified as a risk factor for acute coronary syndromes. We show that GDF-15 expression is up-regulated as disease progresses in murine atherosclerosis and primarily colocalizes with plaque macrophages. Hematopoietic GDF-15 deficiency in low density lipoprotein receptor(-/-) mice led to impaired initial lesion formation and increased collagen in later lesions. Although lesion burden in GDF-15(-/-) chimeras was unaltered, plaques had reduced macrophage infiltrates and decreased necrotic core formation, all features of improved plaque stability. In vitro studies pointed to a TGFβRII-dependent regulatory role of GDF-15 in cell death regulation. Importantly, GDF-15(-/-) macrophages displayed reduced CCR2 expression, whereas GDF-15 promoted macrophage chemotaxis in a strictly CCR2- and TGFβRII-dependent manner, a phenomenon which was not observed in G protein-coupled receptor kinase 2(+/-) macrophages. In conclusion, GDF-15 deletion has a beneficial effect both in early and later atherosclerosis by inhibition of CCR2-mediated chemotaxis and by modulating cell death. Our study is the first to identify GDF-15 as an acute phase modifier of CCR2/TGFβRII-dependent inflammatory responses to vascular injury.
Figures
References
-
- Ait-Oufella H., Pouresmail V., Simon T., Blanc-Brude O., Kinugawa K., Merval R., Offenstadt G., Lesèche G., Cohen P.L., Tedgui A., Mallat Z. 2008. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 28:1429–1431 10.1161/ATVBAHA.108.169078 - DOI - PubMed
-
- Aragay A.M., Mellado M., Frade J.M., Martin A.M., Jimenez-Sainz M.C., Martinez-A C., Mayor F., Jr 1998. Monocyte chemoattractant protein-1-induced CCR2B receptor desensitization mediated by the G protein-coupled receptor kinase 2. Proc. Natl. Acad. Sci. USA. 95:2985–2990 10.1073/pnas.95.6.2985 - DOI - PMC - PubMed
-
- Bootcov M.R., Bauskin A.R., Valenzuela S.M., Moore A.G., Bansal M., He X.Y., Zhang H.P., Donnellan M., Mahler S., Pryor K., et al. 1997. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc. Natl. Acad. Sci. USA. 94:11514–11519 10.1073/pnas.94.21.11514 - DOI - PMC - PubMed
-
- Brown D.A., Moore J., Johnen H., Smeets T.J., Bauskin A.R., Kuffner T., Weedon H., Milliken S.T., Tak P.P., Smith M.D., Breit S.N. 2007. Serum macrophage inhibitory cytokine 1 in rheumatoid arthritis: a potential marker of erosive joint destruction. Arthritis Rheum. 56:753–764 10.1002/art.22410 - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
