

Towards understanding the epigenetics of transcription by chromatin structure and the nuclear matrix
- PMID: 21243045
- PMCID: PMC3021472
Towards understanding the epigenetics of transcription by chromatin structure and the nuclear matrix
Abstract
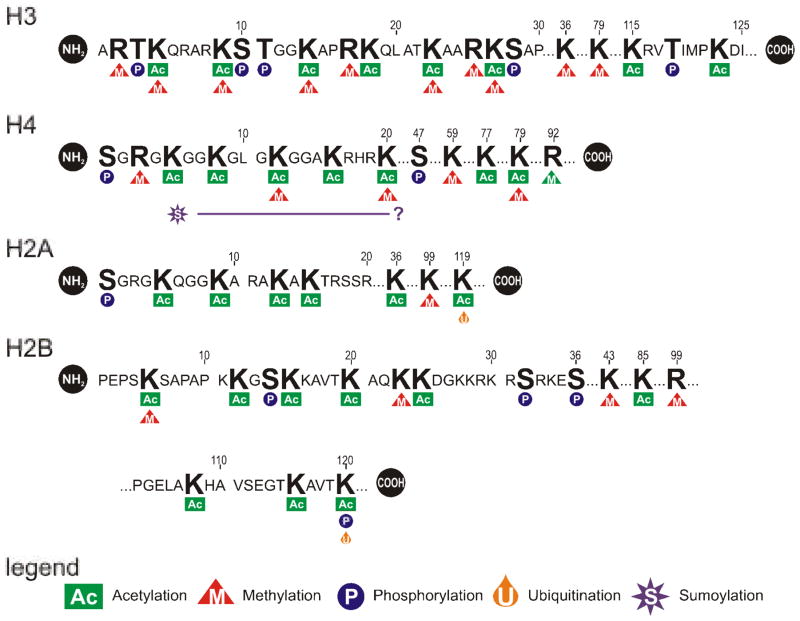
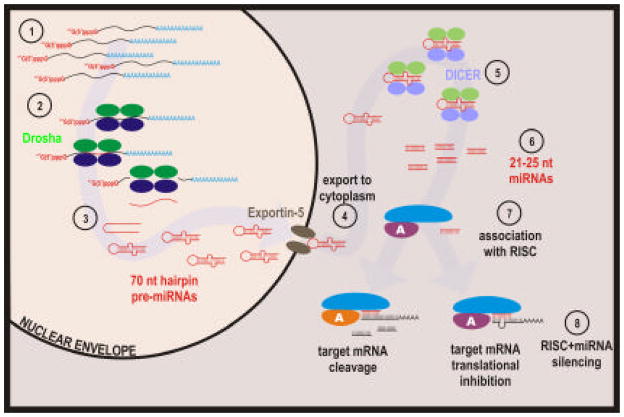
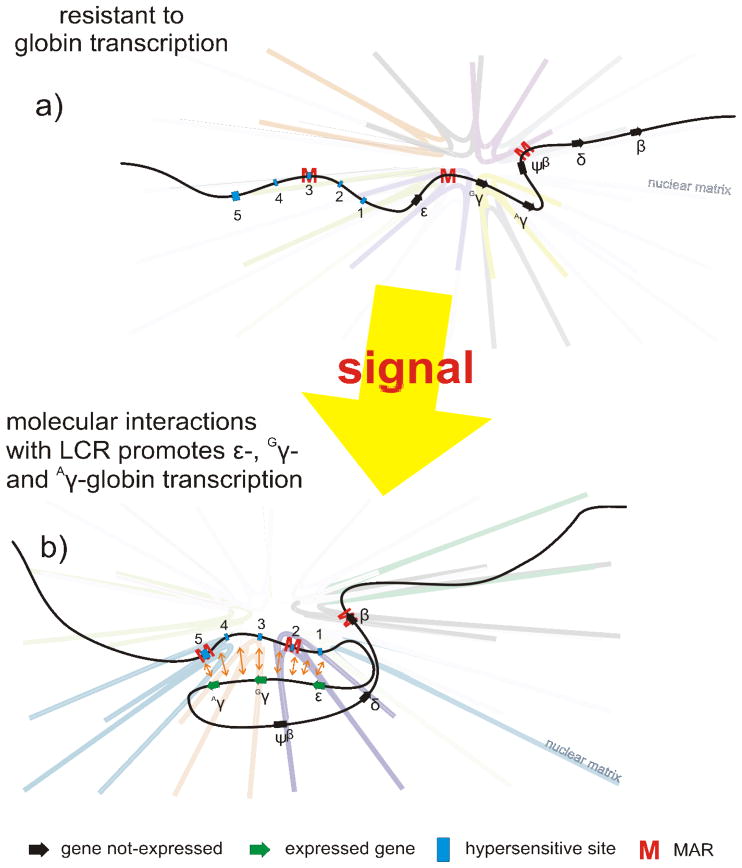
The eukaryotic nucleus houses a significant amount of information that is carefully ordered to ensure that genes can be transcribed as needed throughout development and differentiation. The genome is partitioned into regions containing functional transcription units, providing the means for the cell to selectively activate some, while keeping other regions of the genome silent. Over the last quarter of a century the structure of chromatin and how it is influenced by epigenetics has come into the forefront of modern biology. However, it has thus far failed to identify the mechanism by which individual genes or domains are selected for expression. Through covalent and structural modification of the DNA and chromatin proteins, epigenetics maintains both active and silent chromatin states. This is the "other" genetic code, often superseding that dictated by the nucleotide sequence. The nuclear matrix is rich in many of the factors that govern nuclear processes. It includes a host of unknown factors that may provide our first insight into the structural mechanism responsible for the genetic selectivity of a differentiating cell. This review will consider the nuclear matrix as an integral component of the epigenetic mechanism.
Figures
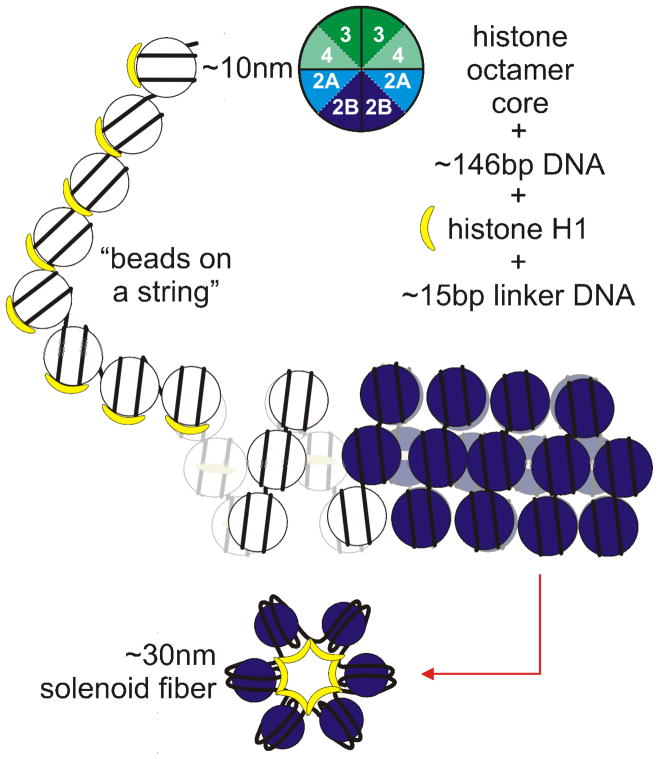
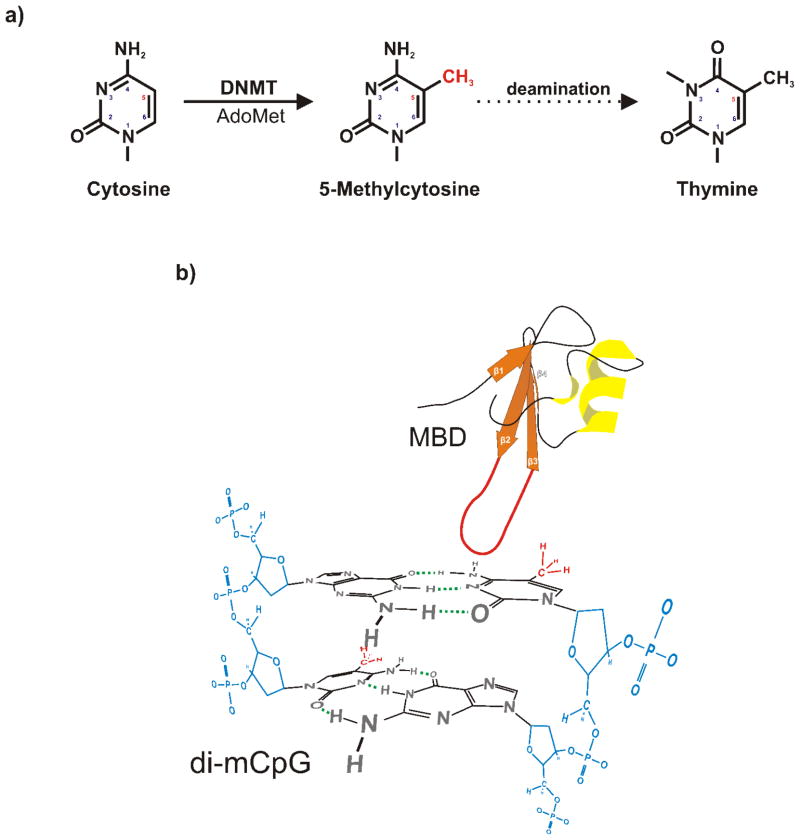
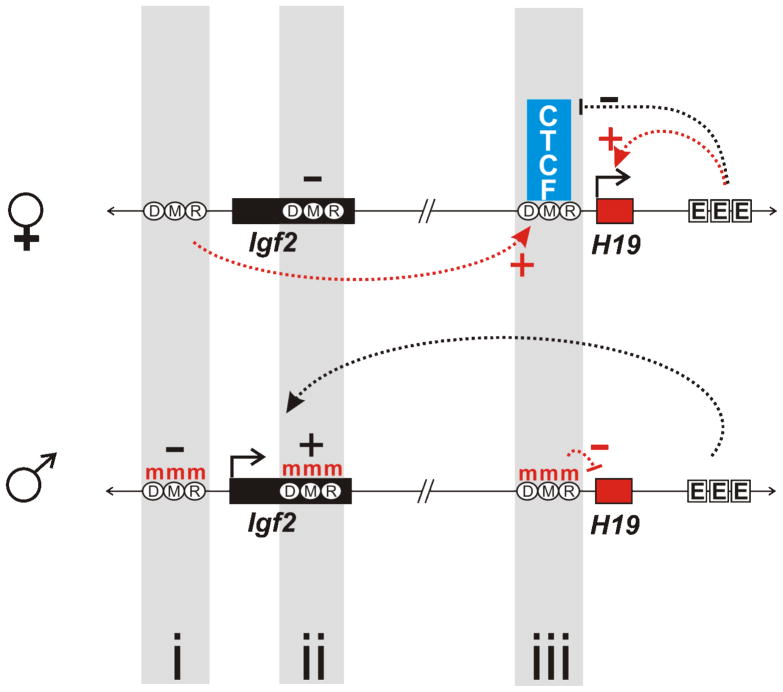
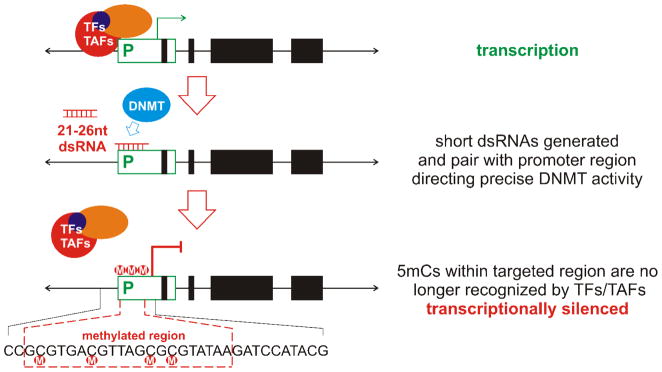

References
-
- Aapola U, Lyle R, Krohn K, Antonarakis SE, Peterson P. Isolation and initial characterization of the mouse Dnmt3l gene. Cytogenet Cell Genet. 2001;92:122–6. - PubMed
-
- Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–92. - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. - PubMed
-
- Amir RE, Zoghbi HY. Rett syndrome: methyl-CpG-binding protein 2 mutations and phenotype-genotype correlations. Am J Med Genet. 2000;97:147–52. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources