

Smad4 inactivation promotes malignancy and drug resistance of colon cancer
- PMID: 21245094
- PMCID: PMC3075468
- DOI: 10.1158/0008-5472.CAN-09-3269
Smad4 inactivation promotes malignancy and drug resistance of colon cancer
Abstract
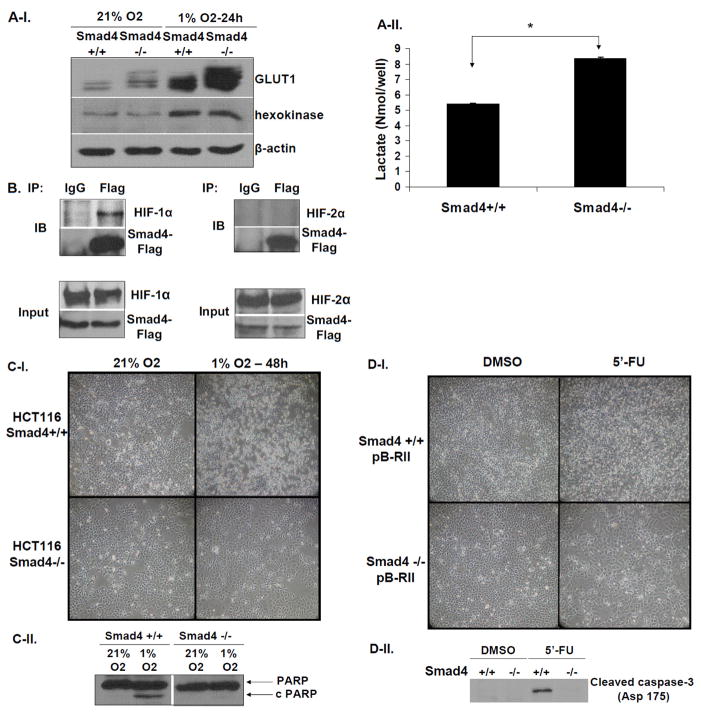
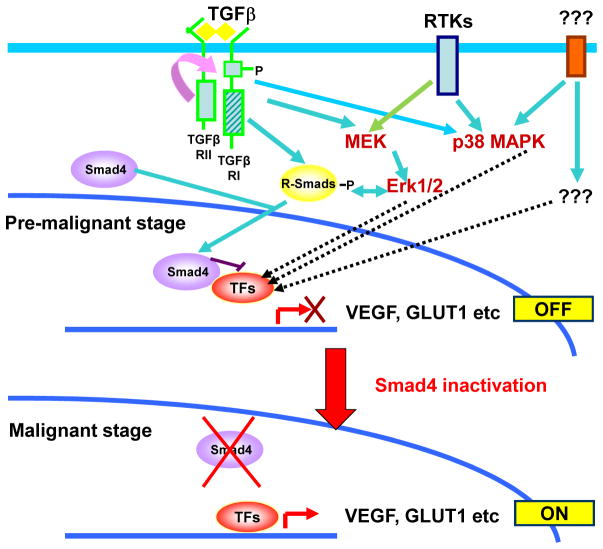
SMAD4 is localized to chromosome 18q21, a frequent site for loss of heterozygosity in advanced stage colon cancers. Although Smad4 is regarded as a signaling mediator of the TGFβ signaling pathway, its role as a major suppressor of colorectal cancer progression and the molecular events underlying this phenomenon remain elusive. Here, we describe the establishment and use of colon cancer cell line model systems to dissect the functional roles of TGFβ and Smad4 inactivation in the manifestation of a malignant phenotype. We found that loss of function of Smad4 and retention of intact TGFβ receptors could synergistically increase the levels of VEGF, a major proangiogenic factor. Pharmacologic inhibition studies suggest that overactivation of the TGFβ-induced MEK-Erk and p38-MAPK (mitogen-activated protein kinase) auxiliary pathways are involved in the induction of VEGF expression in SMAD4 null cells. Overall, SMAD4 deficiency was responsible for the enhanced migration of colon cancer cells with a corresponding increase in matrix metalloprotease 9 enhanced hypoxia-induced GLUT1 expression, increased aerobic glycolysis, and resistance to 5'-fluoruracil-mediated apoptosis. Interestingly, Smad4 specifically interacts with hypoxia-inducible factor (HIF) 1α under hypoxic conditions providing a molecular basis for the differential regulation of target genes to suppress a malignant phenotype. In summary, our results define a molecular mechanism that explains how loss of the tumor suppressor Smad4 promotes colorectal cancer progression. These findings are also consistent with targeting TGFβ-induced auxiliary pathways, such as MEK-ERK, and p38-MAPK and the glycolytic cascade, in SMAD4-deficient tumors as attractive strategies for therapeutic intervention.
Figures
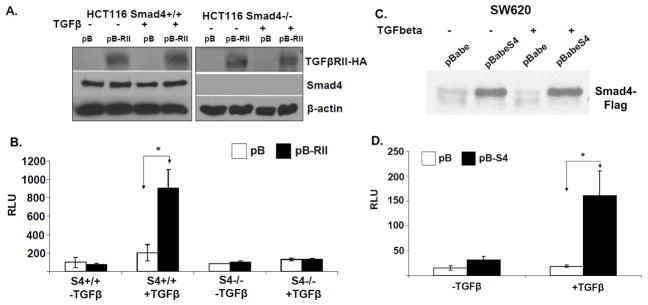
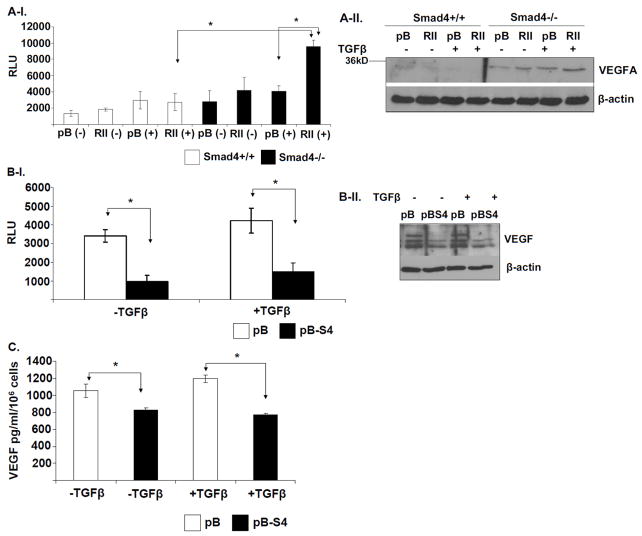
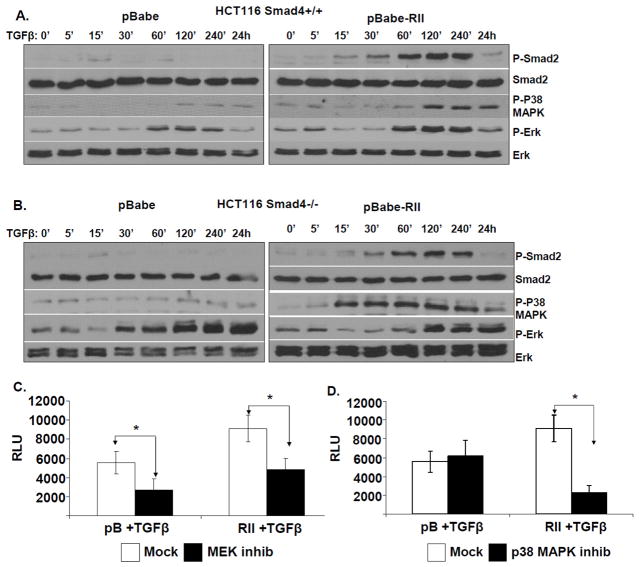
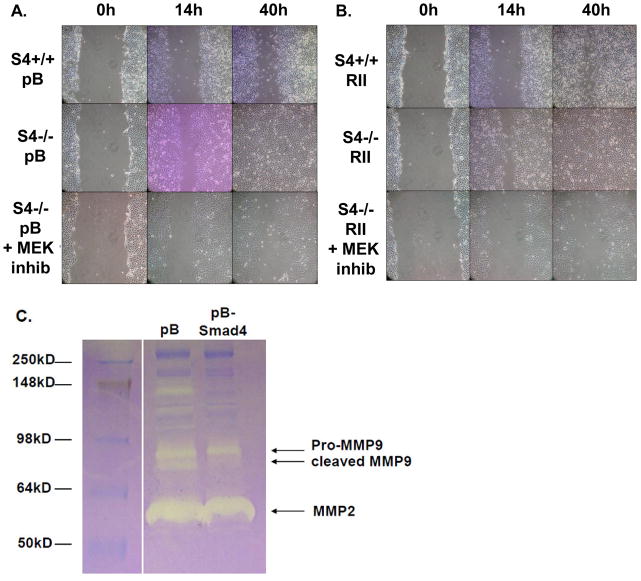
References
-
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–49. - PubMed
-
- Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. - PubMed
-
- Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386:761–63. - PubMed
-
- Thiagalingam S, Cheng K-h, Foy RL, Lee HJ, Chinnappan D, Ponte JF. TGF-beta and its Smad connection to cancer. Current Genomics. 2002;3:449–76.
-
- Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–91. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
