

NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance
- PMID: 21245135
- PMCID: PMC3058977
- DOI: 10.1074/jbc.M110.196790
NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance
Abstract
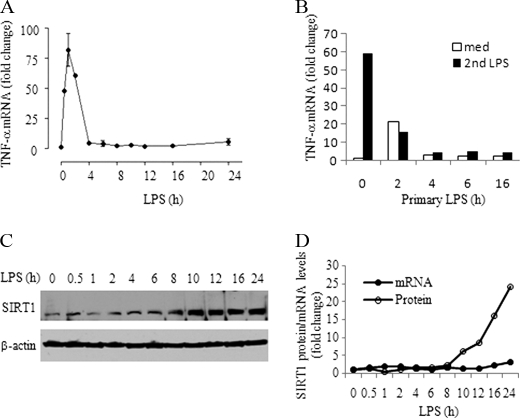
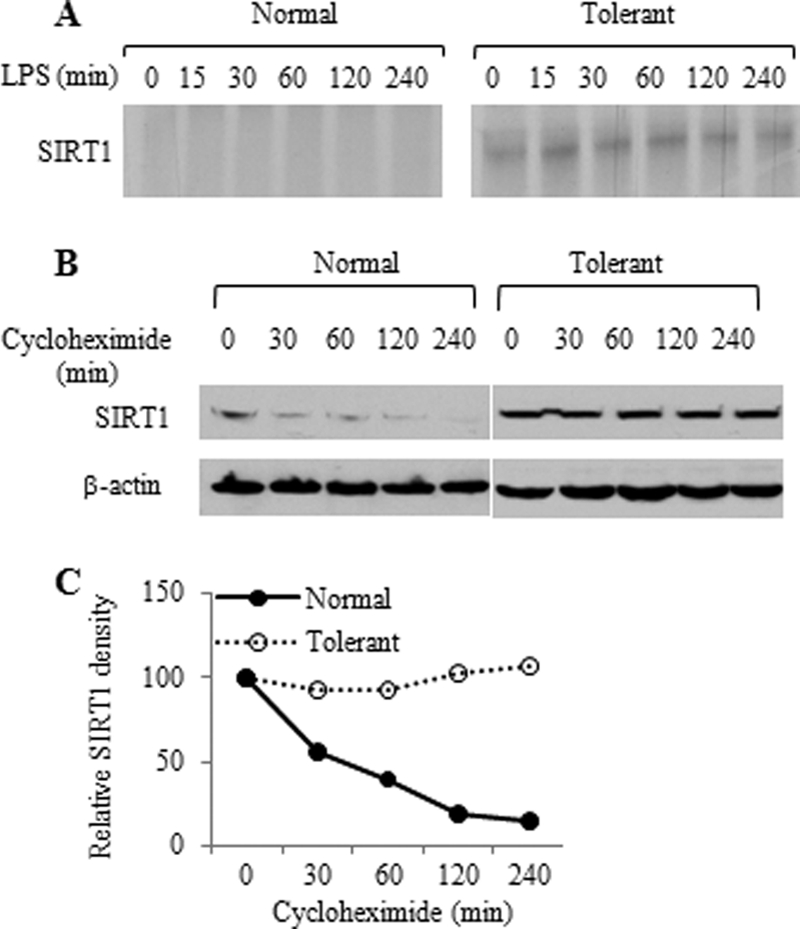
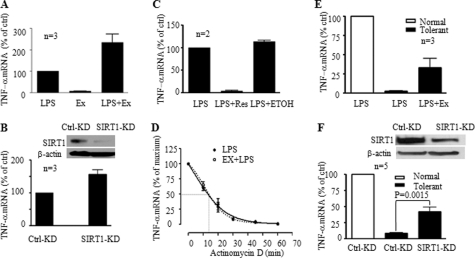
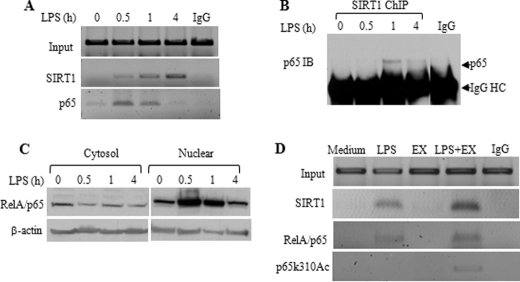
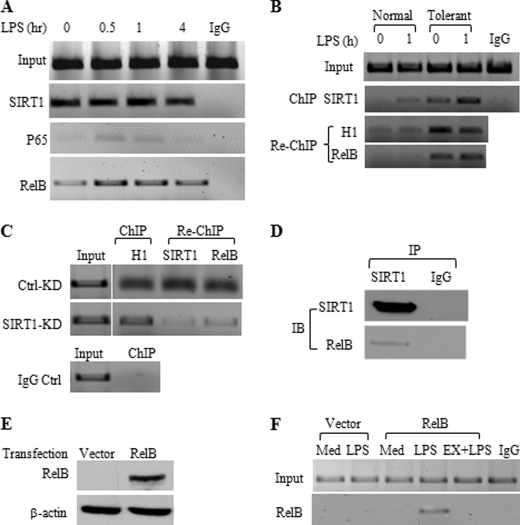
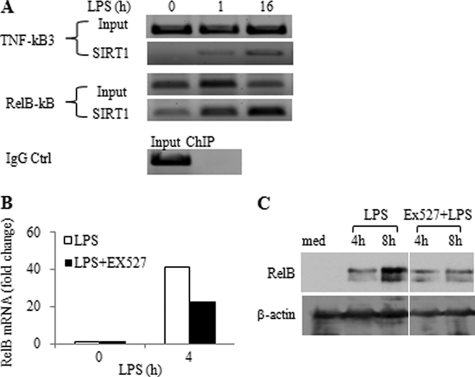
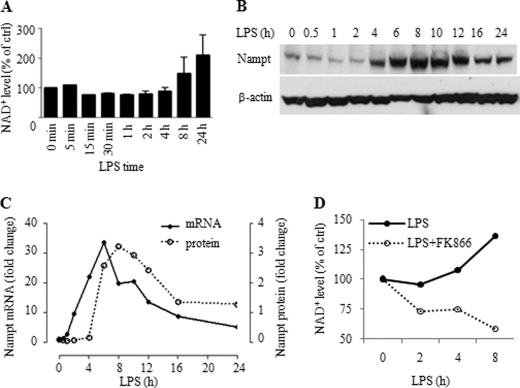
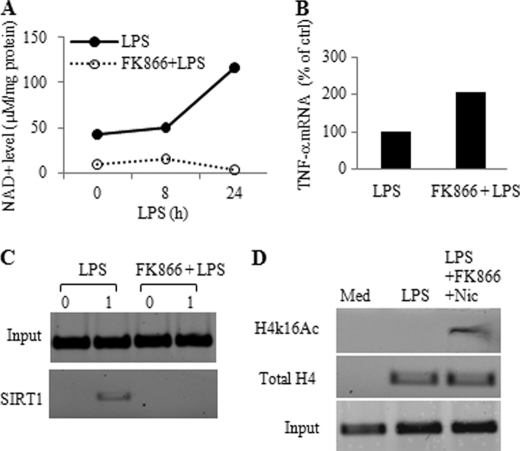
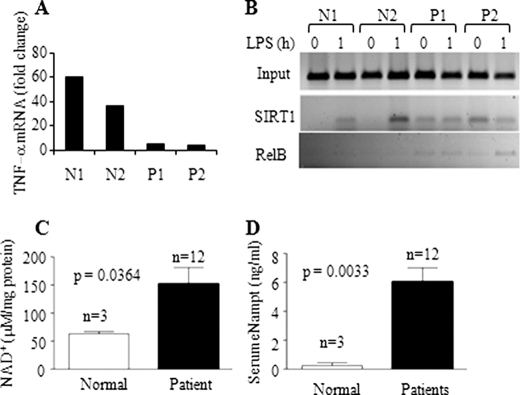
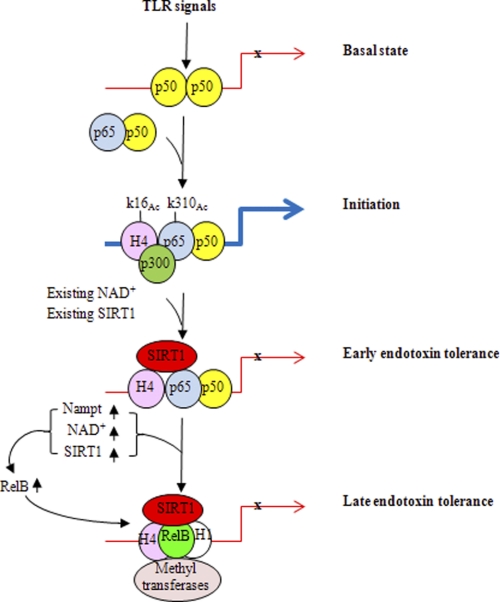
Gene-selective epigenetic reprogramming and shifts in cellular bioenergetics develop when Toll-like receptors (TLR) recognize and respond to systemic life-threatening infections. Using a human monocyte cell model of endotoxin tolerance and human leukocytes from acute systemic inflammation with sepsis, we report that energy sensor sirtuin 1 (SIRT1) coordinates the epigenetic and bioenergy shifts. After TLR4 signaling, SIRT1 rapidly accumulated at the promoters of TNF-α and IL-1β, but not IκBα; SIRT1 promoter binding was dependent on its co-factor, NAD(+). During this initial process, SIRT1 deacetylated RelA/p65 lysine 310 and nucleosomal histone H4 lysine 16 to promote termination of NFκB-dependent transcription. SIRT1 then remained promoter bound and recruited de novo induced RelB, which directed assembly of the mature transcription repressor complex that generates endotoxin tolerance. SIRT1 also promoted de novo expression of RelB. During sustained endotoxin tolerance, nicotinamide phosphoribosyltransferase (Nampt), the rate-limiting enzyme for endogenous production of NAD(+), and SIRT1 expression increased. The elevation of SIRT1 required protein stabilization and enhanced translation. To support the coordination of bioenergetics in human sepsis, we observed elevated NAD(+) levels concomitant with SIRT1 and RelB accumulation at the TNF-α promoter of endotoxin tolerant sepsis blood leukocytes. We conclude that TLR4 stimulation and human sepsis activate pathways that couple NAD(+) and its sensor SIRT1 with epigenetic reprogramming.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
