

Proteome-wide systems analysis of a cellulosic biofuel-producing microbe
- PMID: 21245846
- PMCID: PMC3049413
- DOI: 10.1038/msb.2010.116
Proteome-wide systems analysis of a cellulosic biofuel-producing microbe
Abstract
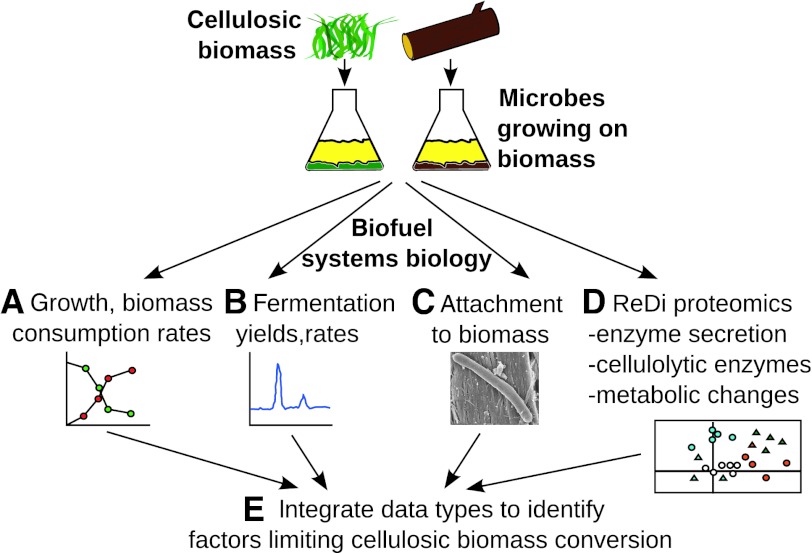
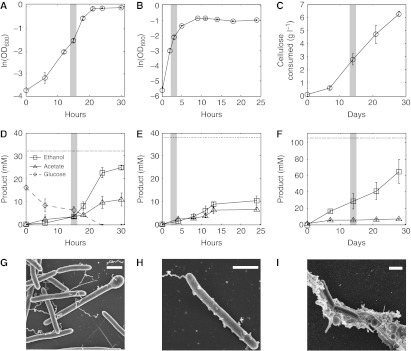
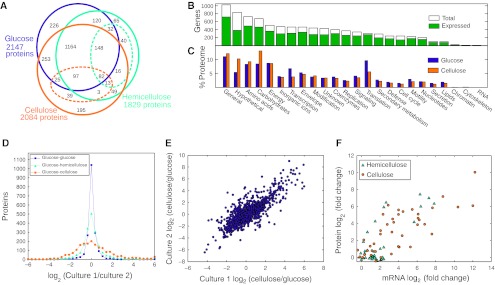
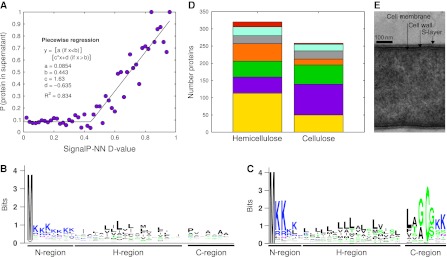
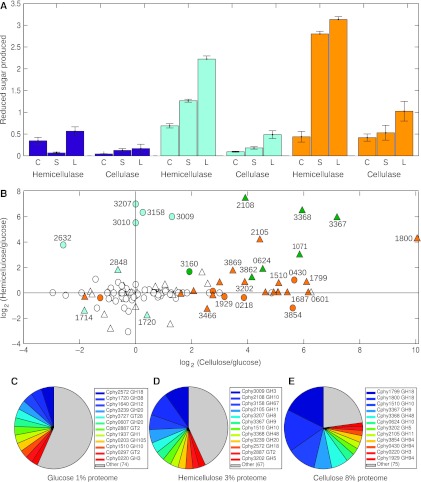
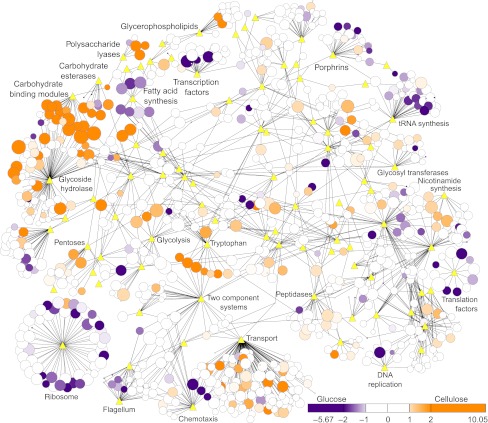
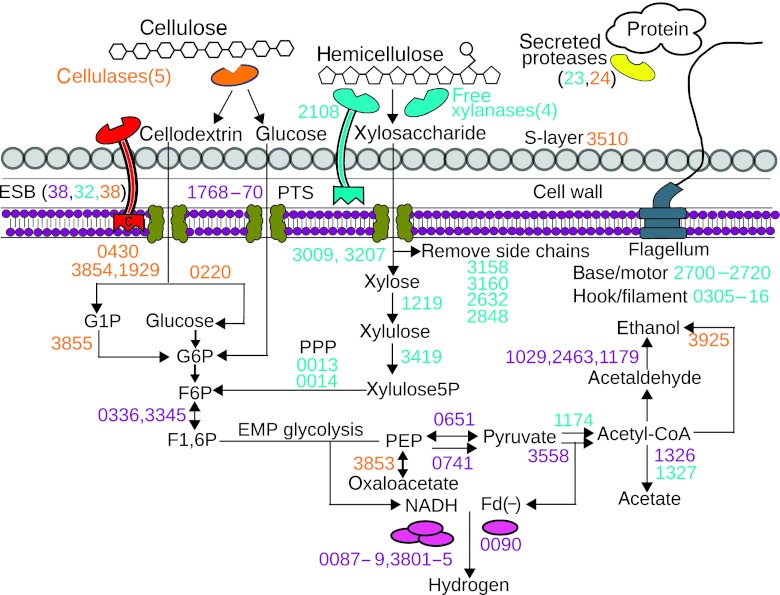
Fermentation of plant biomass by microbes like Clostridium phytofermentans recycles carbon globally and can make biofuels from inedible feedstocks. We analyzed C. phytofermentans fermenting cellulosic substrates by integrating quantitative mass spectrometry of more than 2500 proteins with measurements of growth, enzyme activities, fermentation products, and electron microscopy. Absolute protein concentrations were estimated using Absolute Protein EXpression (APEX); relative changes between treatments were quantified with chemical stable isotope labeling by reductive dimethylation (ReDi). We identified the different combinations of carbohydratases used to degrade cellulose and hemicellulose, many of which were secreted based on quantification of supernatant proteins, as well as the repertoires of glycolytic enzymes and alcohol dehydrogenases (ADHs) enabling ethanol production at near maximal yields. Growth on cellulose also resulted in diverse changes such as increased expression of tryptophan synthesis proteins and repression of proteins for fatty acid metabolism and cell motility. This study gives a systems-level understanding of how this microbe ferments biomass and provides a rational, empirical basis to identify engineering targets for industrial cellulosic fermentation.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Bendtsen JD, Nielsen H, von Heijne G, Brunak S (2004) Improved prediction of signal peptides: SignalP 3.0. J Mol Biol 340: 783–795 - PubMed
-
- Blouzard JC, Coutinho PM, Fierobe HP, Henrissat B, Lignon S, Tardif C, Pagès S, de Philip P (2010) Modulation of cellulosome composition in Clostridium cellulolyticum: adaptation to the polysaccharide environment revealed by proteomic and carbohydrate-active enzyme analyses. Proteomics 10: 541–554 - PubMed
-
- Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJR (2009) Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4: 484–494 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
