

Crystal structure of a bacterial phosphoglucomutase, an enzyme involved in the virulence of multiple human pathogens
- PMID: 21246636
- PMCID: PMC3066478
- DOI: 10.1002/prot.22957
Crystal structure of a bacterial phosphoglucomutase, an enzyme involved in the virulence of multiple human pathogens
Abstract
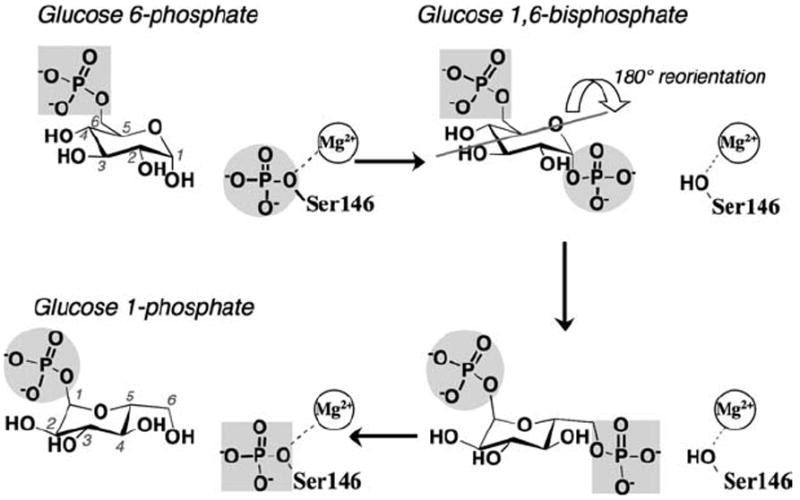
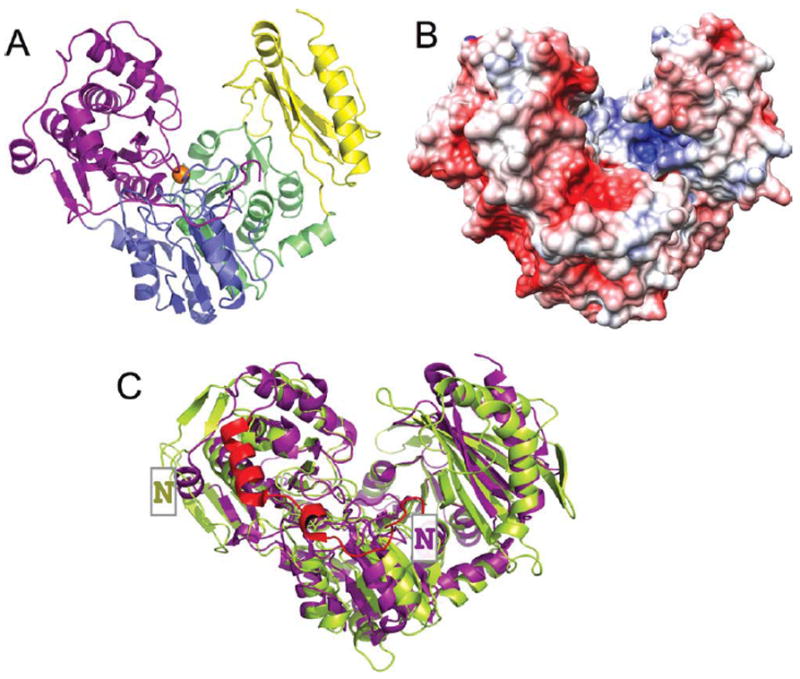
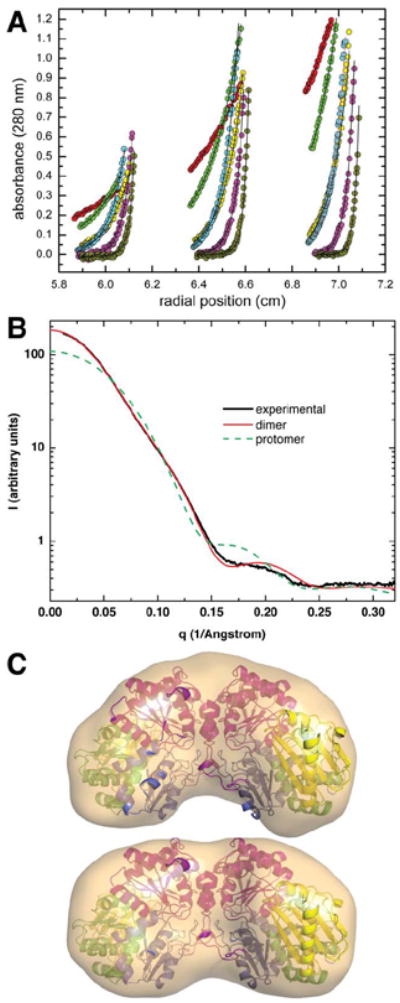
The crystal structure of the enzyme phosphoglucomutase from Salmonella typhimurium (StPGM) is reported at 1.7 A resolution. This is the first high-resolution structural characterization of a bacterial protein from this large enzyme family, which has a central role in metabolism and is also important to bacterial virulence and infectivity. A comparison of the active site of StPGM with that of other phosphoglucomutases reveals conserved residues that are likely involved in catalysis and ligand binding for the entire enzyme family. An alternate crystal form of StPGM and normal mode analysis give insights into conformational changes of the C-terminal domain that occur upon ligand binding. A novel observation from the StPGM structure is an apparent dimer in the asymmetric unit of the crystal, mediated largely through contacts in an N-terminal helix. Analytical ultracentrifugation and small-angle X-ray scattering confirm that StPGM forms a dimer in solution. Multiple sequence alignments and phylogenetic studies show that a distinct subset of bacterial PGMs share the signature dimerization helix, while other bacterial and eukaryotic PGMs are likely monomers. These structural, biochemical, and bioinformatic studies of StPGM provide insights into the large α-D-phosphohexomutase enzyme superfamily to which it belongs, and are also relevant to the design of inhibitors specific to the bacterial PGMs.
Copyright © 2010 Wiley-Liss, Inc.
Figures


References
-
- Paterson GK, Cone DB, Peters SE, Maskell DJ. The enzyme phosphoglucomutase (Pgm) is required by Salmonella enterica serovar Typhimurium for O-antigen production, resistance to antimicrobial peptides and in vivo fitness. Microbiology. 2009;155(Part 10):3403–3410. - PubMed
-
- Bizzini A, Majcherczyk P, Beggah-Moller S, Soldo B, Entenza JM, Gaillard M, Moreillon P, Lazarevic V. Effects of alpha-phosphoglucomutase deficiency on cell wall properties and fitness in Streptococcus gordonii. Microbiology. 2007;153(Part 2):490–498. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
