

Prion propagation in cells expressing PrP glycosylation mutants
- PMID: 21248032
- PMCID: PMC3067877
- DOI: 10.1128/JVI.02257-10
Prion propagation in cells expressing PrP glycosylation mutants
Abstract
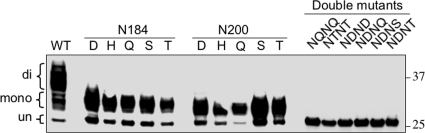
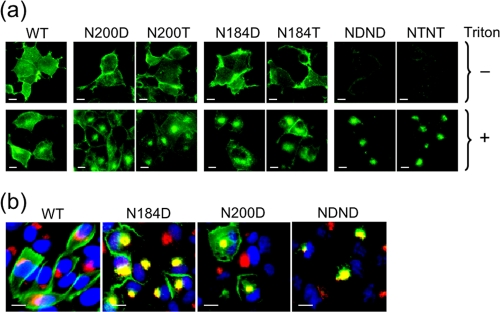
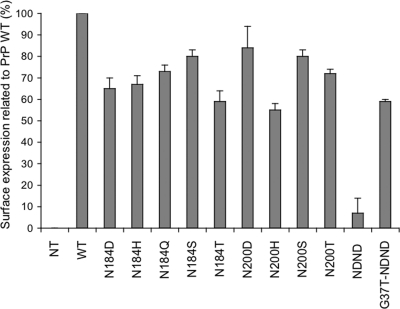
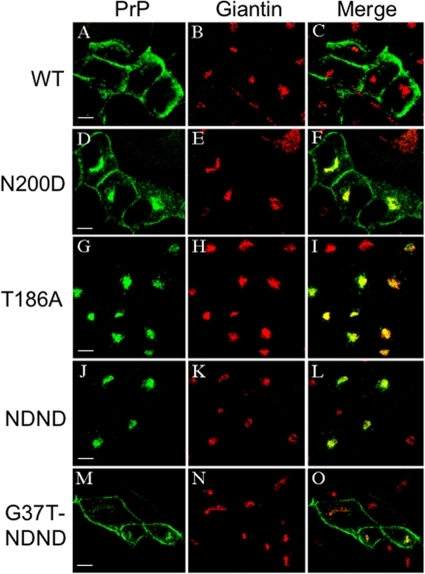
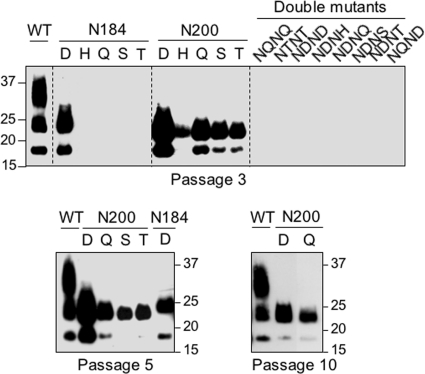
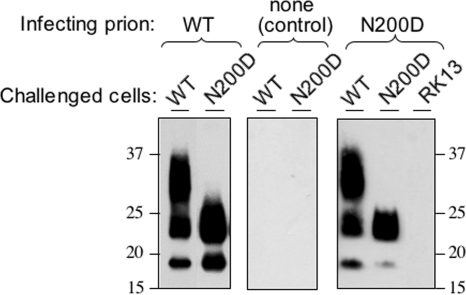
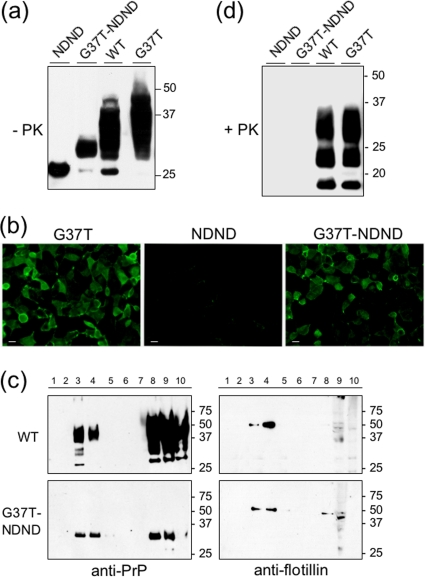
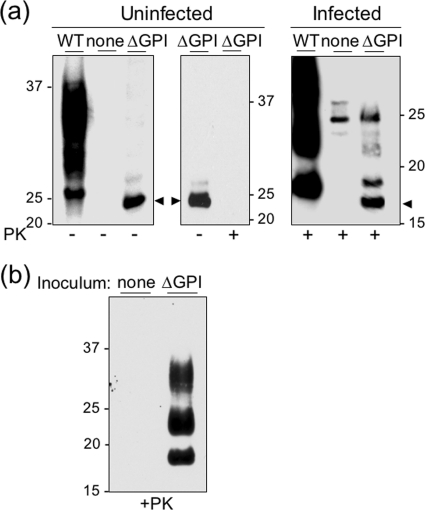
Infection by prions involves conversion of a host-encoded cell surface protein (PrP(C)) to a disease-related isoform (PrP(Sc)). PrP(C) carries two glycosylation sites variably occupied by complex N-glycans, which have been suggested by previous studies to influence the susceptibility to these diseases and to determine characteristics of prion strains. We used the Rov cell system, which is susceptible to sheep prions, to generate a series of PrP(C) glycosylation mutants with mutations at one or both attachment sites. We examined their subcellular trafficking and ability to convert into PrP(Sc) and to sustain stable prion propagation in the absence of wild-type PrP. The susceptibility to infection of mutants monoglycosylated at either site differed dramatically depending on the amino acid substitution. Aglycosylated double mutants showed overaccumulation in the Golgi compartment and failed to be infected. Introduction of an ectopic glycosylation site near the N terminus fully restored cell surface expression of PrP but not convertibility into PrP(Sc), while PrP(C) with three glycosylation sites conferred cell permissiveness to infection similarly to the wild type. In contrast, predominantly aglycosylated molecules with nonmutated N-glycosylation sequons, produced in cells expressing glycosylphosphatidylinositol-anchorless PrP(C), were able to form infectious PrP(Sc). Together our findings suggest that glycosylation is important for efficient trafficking of anchored PrP to the cell surface and sustained prion propagation. However, properly trafficked glycosylation mutants were not necessarily prone to conversion, thus making it difficult in such studies to discern whether the amino acid changes or glycan chain removal most influences the permissiveness to prion infection.
Figures
References
-
- Aguzzi, A., F. Baumann, and J. Bremer. 2008. The prion's elusive reason for being. Annu. Rev. Neurosci. 31:439-477. - PubMed
-
- Beringue, V., et al. 2003. Regional heterogeneity of cellular prion protein isoforms in the mouse brain. Brain 126:2065-2073. - PubMed
-
- Beringue, V., J. L. Vilotte, and H. Laude. 2008. Prion agent diversity and species barrier. Vet. Res. 39:47. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
