

The phenotype of recurrent 10q22q23 deletions and duplications
- PMID: 21248748
- PMCID: PMC3060324
- DOI: 10.1038/ejhg.2010.211
The phenotype of recurrent 10q22q23 deletions and duplications
Abstract
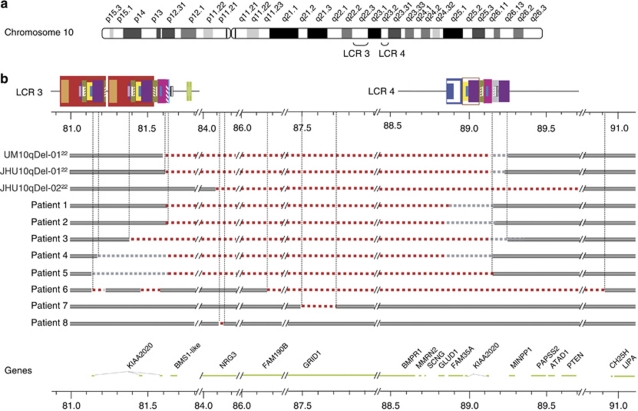
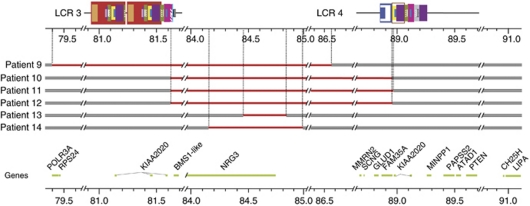
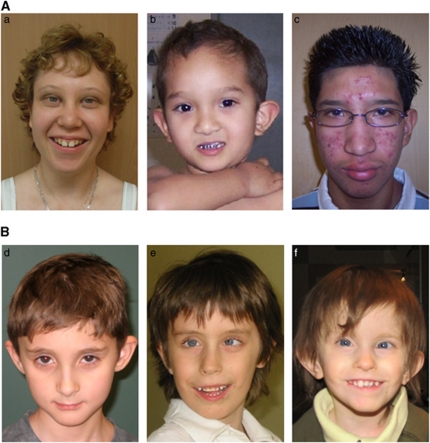
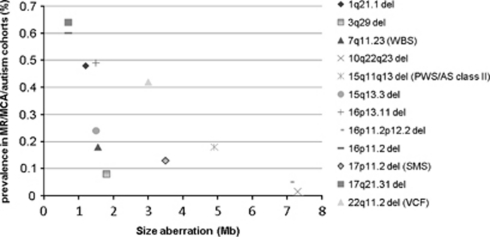
The genomic architecture of the 10q22q23 region is characterised by two low-copy repeats (LCRs3 and 4), and deletions in this region appear to be rare. We report the clinical and molecular characterisation of eight novel deletions and six duplications within the 10q22.3q23.3 region. Five deletions and three duplications occur between LCRs3 and 4, whereas three deletions and three duplications have unique breakpoints. Most of the individuals with the LCR3-4 deletion had developmental delay, mainly affecting speech. In addition, macrocephaly, mild facial dysmorphisms, cerebellar anomalies, cardiac defects and congenital breast aplasia were observed. For congenital breast aplasia, the NRG3 gene, known to be involved in early mammary gland development in mice, is a putative candidate gene. For cardiac defects, BMPR1A and GRID1 are putative candidate genes because of their association with cardiac structure and function. Duplications between LCRs3 and 4 are associated with variable phenotypic penetrance. Probands had speech and/or motor delays and dysmorphisms including a broad forehead, deep-set eyes, upslanting palpebral fissures, a smooth philtrum and a thin upper lip. In conclusion, duplications between LCRs3 and 4 on 10q22.3q23.2 may lead to a distinct facial appearance and delays in speech and motor development. However, the phenotypic spectrum is broad, and duplications have also been found in healthy family members of a proband. Reciprocal deletions lead to speech and language delay, mild facial dysmorphisms and, in some individuals, to cerebellar, breast developmental and cardiac defects.
Figures
References
-
- Schmickel RD. Contiguous gene syndromes: a component of recognizable syndromes. J Pediatr. 1986;109:231–241. - PubMed
-
- Ewart AK, Morris CA, Atkinson D, et al. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. - PubMed
-
- Bowen P, Biederman B, Hoo JJ. The critical segment for the Langer-Giedion syndrome: 8q24.11----q24.12. Ann Genet. 1985;28:224–227. - PubMed
-
- Ledbetter DH, Riccardi VM, Airhart SD, et al. Deletions of chromosome 15 as a cause of the Prader-Willi syndrome. N Engl J Med. 1981;304:325–329. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
