

Wilms' tumours: about tumour suppressor genes, an oncogene and a chameleon gene
- PMID: 21248786
- PMCID: PMC4332715
- DOI: 10.1038/nrc3002
Wilms' tumours: about tumour suppressor genes, an oncogene and a chameleon gene
Abstract
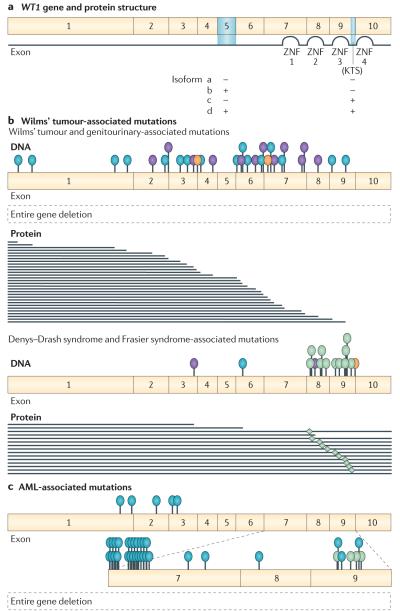
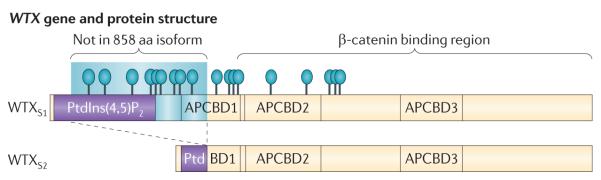
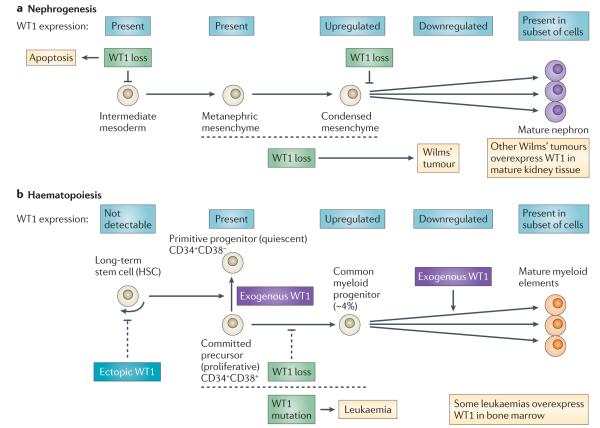
Genes identified as being mutated in Wilms' tumour include TP53, a classic tumour suppressor gene (TSG); CTNNB1 (encoding β-catenin), a classic oncogene; WTX, which accumulating data indicate is a TSG; and WT1, which is inactivated in some Wilms' tumours, similar to a TSG. However, WT1 does not always conform to the TSG label, and some data indicate that WT1 enhances cell survival and proliferation, like an oncogene. Is WT1 a chameleon, functioning as either a TSG or an oncogene, depending on cellular context? Are these labels even appropriate for describing and understanding the function of WT1?
Figures
References
-
- Bardeesy N, et al. Anaplastic Wilms’ tumour, a subtype displaying poor prognosis, harbours p53 gene mutations. Nature Genet. 1994;7:91–97. - PubMed
-
-
Huff V. Wilms tumor genetics. Am. J. Med. Genet. 1998;79:260–267. Although an older paper, this still provides a good basic summary of Wilms’ tumour genetics along with primary data and an overall description of the type of WT1 mutations observed in patients with Wilms’ tumour and their tumours that is still valid.
-
-
- Koesters R, et al. Mutational activation of the β-catenin proto-oncogene is a common event in the development of Wilms’ tumors. Cancer Res. 1999;59:3880–3882. - PubMed
-
-
Rivera MN, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science. 2007;315:642–645. This is the original report identifying WTX as a Wilms’ tumour gene. It presents a nice story of going from observing a gene copy number change over a very short genomic region to identifying a new cancer-related gene.
-
-
-
Ruteshouser EC, Robinson SM, Huff V. Wilms tumor genetics: mutations in WT1, WTX, and CTNNB1 account for only about one-third of tumors. Genes Chromosomes Cancer. 2008;47:461–470. This paper presents data from more than 100 Wilms’ tumours regarding the type, frequency and co-occurrence of WT1, WTX and CTNNB1 mutations.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
