

Nationwide molecular surveillance of pandemic H1N1 influenza A virus genomes: Canada, 2009
- PMID: 21249207
- PMCID: PMC3017559
- DOI: 10.1371/journal.pone.0016087
Nationwide molecular surveillance of pandemic H1N1 influenza A virus genomes: Canada, 2009
Abstract
Background: In April 2009, a novel triple-reassortant swine influenza A H1N1 virus ("A/H1N1pdm"; also known as SOIV) was detected and spread globally as the first influenza pandemic of the 21(st) century. Sequencing has since been conducted at an unprecedented rate globally in order to monitor the diversification of this emergent virus and to track mutations that may affect virus behavior.
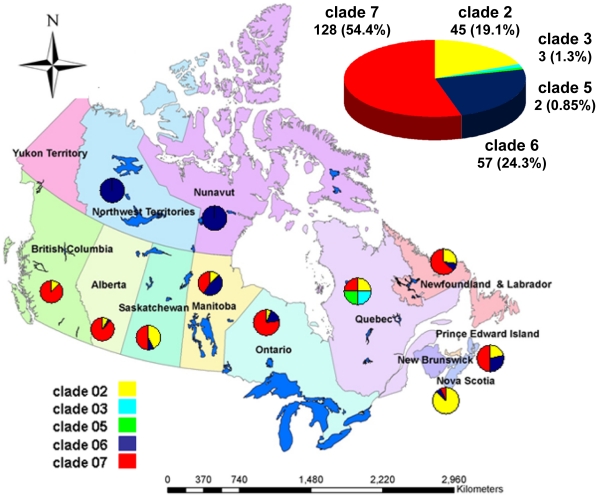
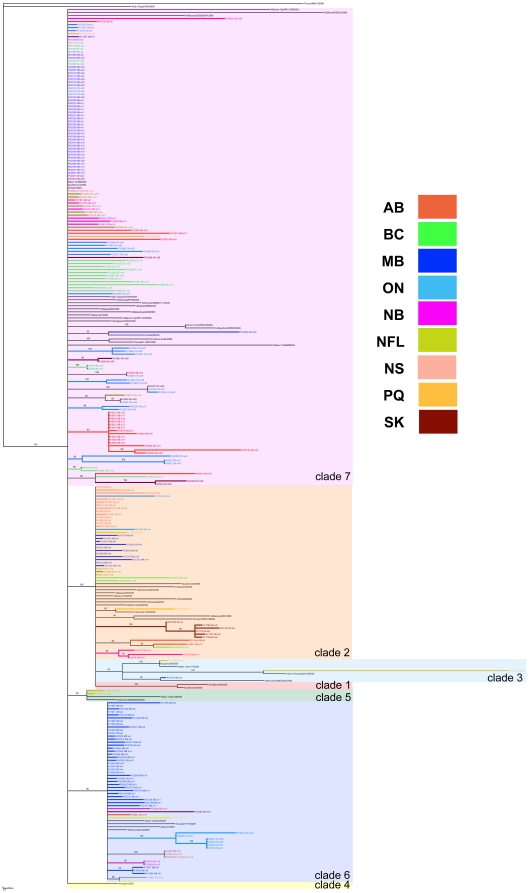
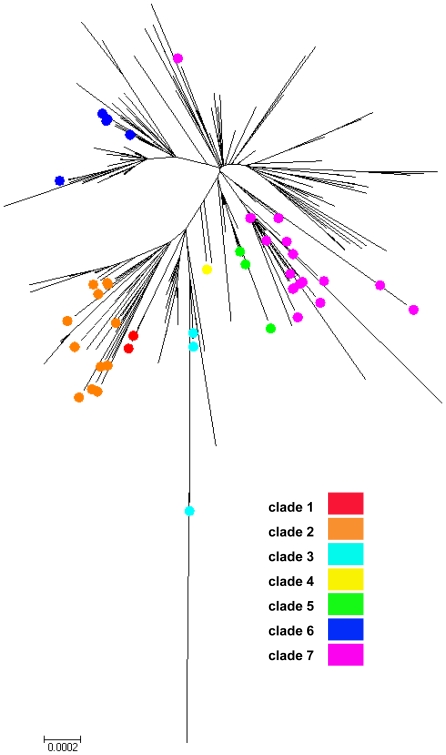
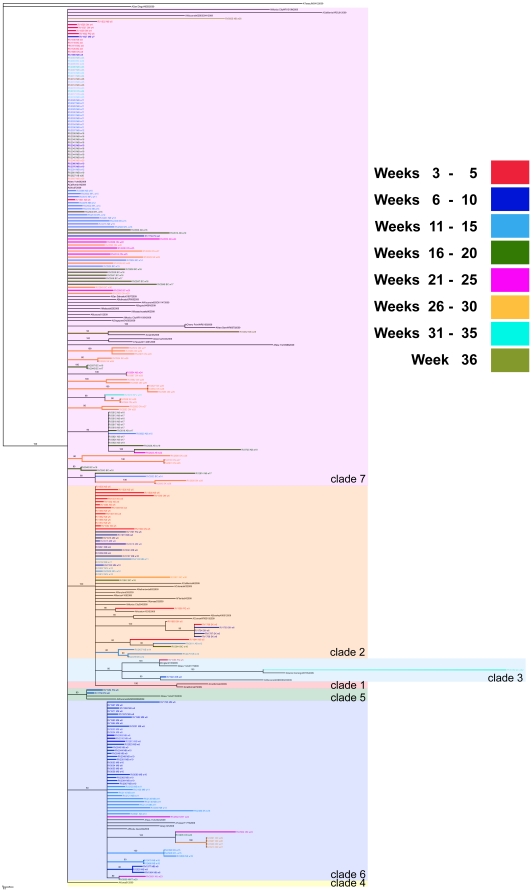
Methodology/principal findings: By Sanger sequencing, we determined consensus whole-genome sequences for A/H1N1pdm viruses sampled nationwide in Canada over 33 weeks during the 2009 first and second pandemic waves. A total of 235 virus genomes sampled from unique subjects were analyzed, providing insight into the temporal and spatial trajectory of A/H1N1pdm lineages within Canada. Three clades (2, 3, and 7) were identifiable within the first two weeks of A/H1N1pdm appearance, with clades 5 and 6 appearing thereafter; further diversification was not apparent. Only two viral sites displayed evidence of adaptive evolution, located in hemagglutinin (HA) corresponding to D222 in the HA receptor-binding site, and to E374 at HA2-subunit position 47. Among the Canadian sampled viruses, we observed notable genetic diversity (1.47 x 10⁻³ amino acid substitutions per site) in the gene encoding PB1, particularly within the viral genomic RNA (vRNA)-binding domain (residues 493-757). This genome data set supports the conclusion that A/H1N1pdm is evolving but not excessively relative to other H1N1 influenza A viruses. Entropy analysis was used to investigate whether any mutated A/H1N1pdm protein residues were associated with infection severity; however no virus genotypes were observed to trend with infection severity. One virus that harboured heterozygote coding mutations, including PB2 D567D/G, was attributed to a severe and potentially mixed infection; yet the functional significance of this PB2 mutation remains unknown.
Conclusions/significance: These findings contribute to enhanced understanding of Influenza A/H1N1pdm viral dynamics.
Conflict of interest statement
Figures
References
-
- World Health Organization. Influenza-like illness in the United States and Mexico, 24 April 2009. 2010(8 June): 1. 24 April 2009. 2009. Available: http://www.who.int/csr/don/2009_04_24/en/index.html via the Internet. 8 June 2010.
-
- Bautista E, Chotpitayasunondh T, Gao Z, Harper SA, et al. Writing Committee of the WHO Consultation on Clinical Aspects of Pandemic (H1N1) 2009 Influenza. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N Engl J Med. 2010;362(18):1708–1719. 10.1056/NEJMra1000449. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
