

DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines
- PMID: 21251332
- PMCID: PMC3091299
- DOI: 10.1186/gb-2011-12-1-r10
DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines
Erratum in
- Genome Biol. 2011;12(6):405
Abstract
Background: DNA methylation is an essential epigenetic mechanism involved in gene regulation and disease, but little is known about the mechanisms underlying inter-individual variation in methylation profiles. Here we measured methylation levels at 22,290 CpG dinucleotides in lymphoblastoid cell lines from 77 HapMap Yoruba individuals, for which genome-wide gene expression and genotype data were also available.
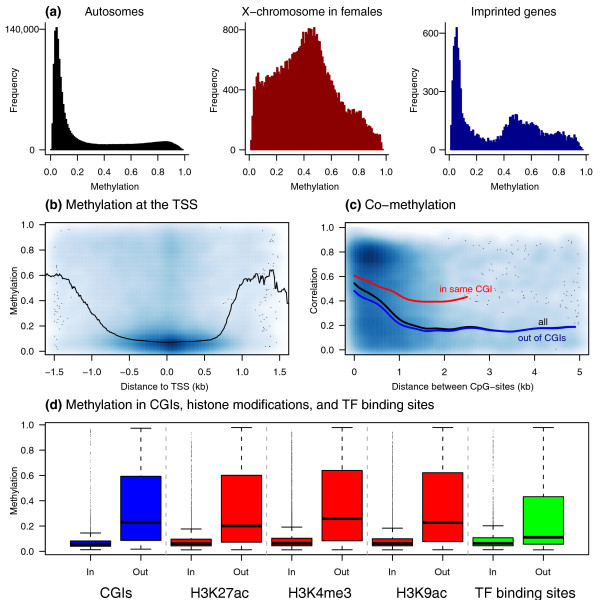
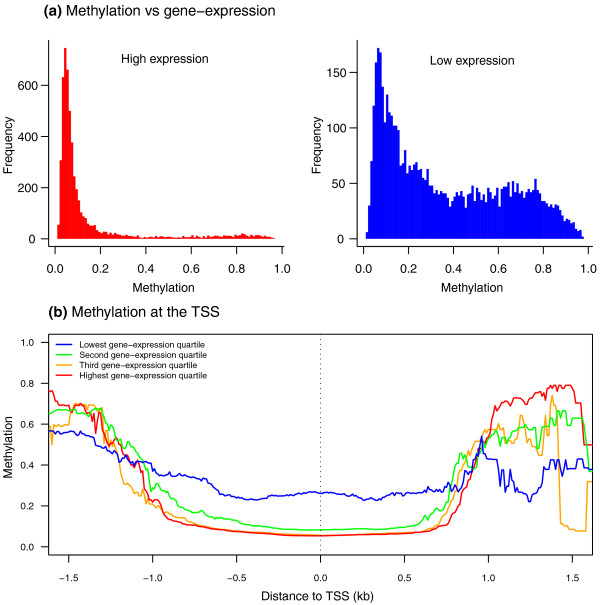
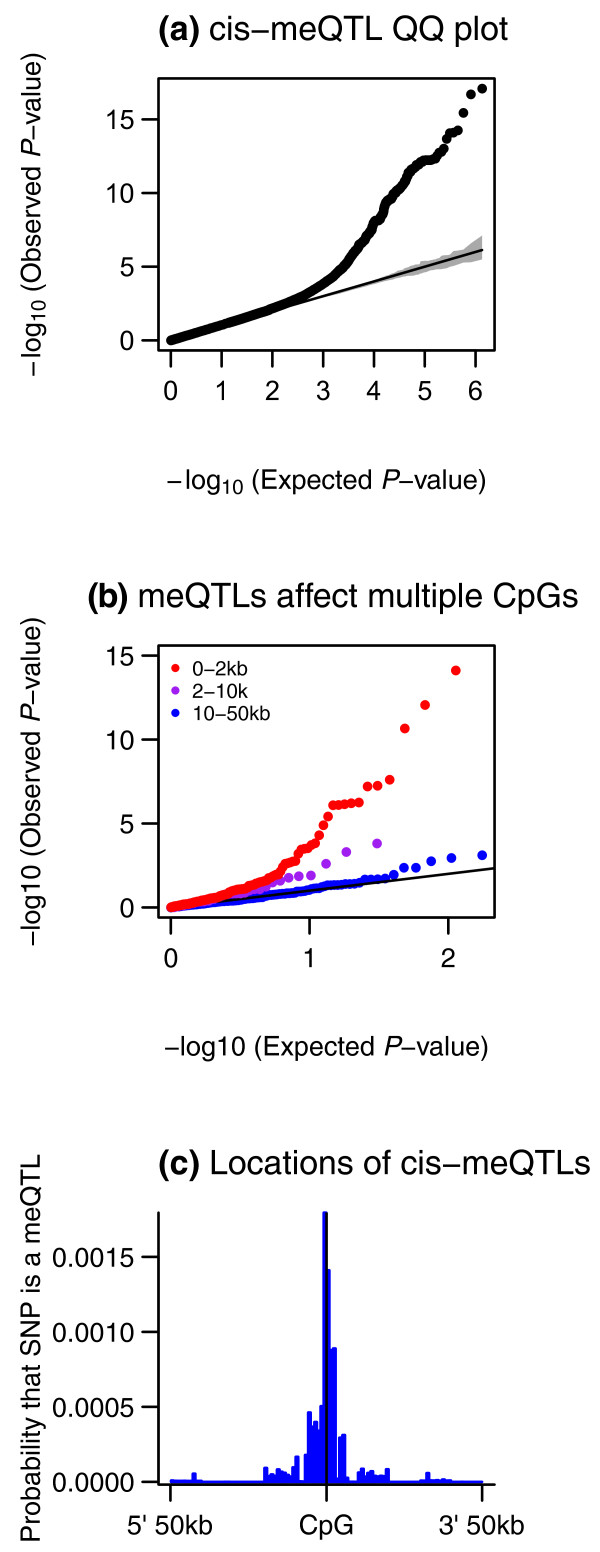
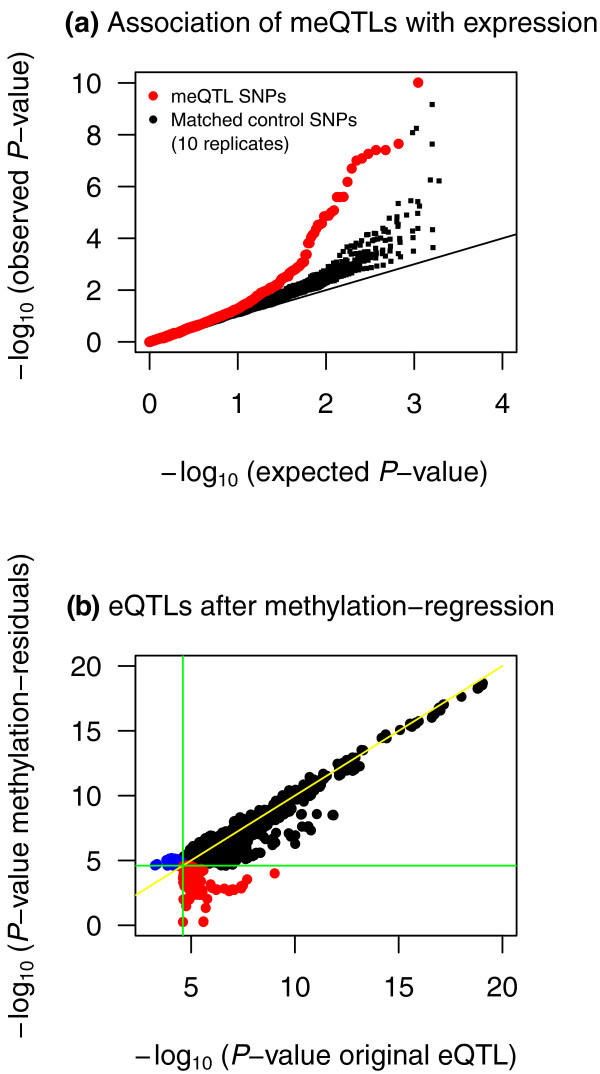
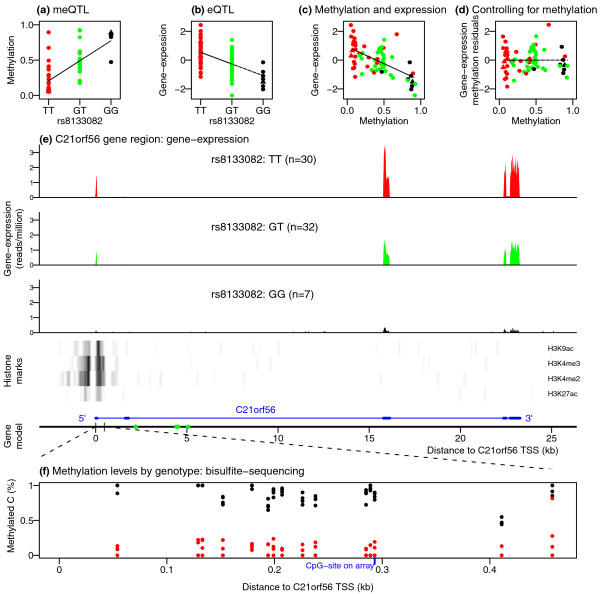
Results: Association analyses of methylation levels with more than three million common single nucleotide polymorphisms (SNPs) identified 180 CpG-sites in 173 genes that were associated with nearby SNPs (putatively in cis, usually within 5 kb) at a false discovery rate of 10%. The most intriguing trans signal was obtained for SNP rs10876043 in the disco-interacting protein 2 homolog B gene (DIP2B, previously postulated to play a role in DNA methylation), that had a genome-wide significant association with the first principal component of patterns of methylation; however, we found only modest signal of trans-acting associations overall. As expected, we found significant negative correlations between promoter methylation and gene expression levels measured by RNA-sequencing across genes. Finally, there was a significant overlap of SNPs that were associated with both methylation and gene expression levels.
Conclusions: Our results demonstrate a strong genetic component to inter-individual variation in DNA methylation profiles. Furthermore, there was an enrichment of SNPs that affect both methylation and gene expression, providing evidence for shared mechanisms in a fraction of genes.
Figures
References
-
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. - DOI - PMC - PubMed
-
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. - DOI - PMC - PubMed
-
- Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. - DOI - PMC - PubMed
-
- Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB. Abundant quantitative trait Loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
