

The MRE11 complex: starting from the ends
- PMID: 21252998
- PMCID: PMC3905242
- DOI: 10.1038/nrm3047
The MRE11 complex: starting from the ends
Abstract
The maintenance of genome stability depends on the DNA damage response (DDR), which is a functional network comprising signal transduction, cell cycle regulation and DNA repair. The metabolism of DNA double-strand breaks governed by the DDR is important for preventing genomic alterations and sporadic cancers, and hereditary defects in this response cause debilitating human pathologies, including developmental defects and cancer. The MRE11 complex, composed of the meiotic recombination 11 (MRE11), RAD50 and Nijmegen breakage syndrome 1 (NBS1; also known as nibrin) proteins is central to the DDR, and recent insights into its structure and function have been gained from in vitro structural analysis and studies of animal models in which the DDR response is deficient.
Figures
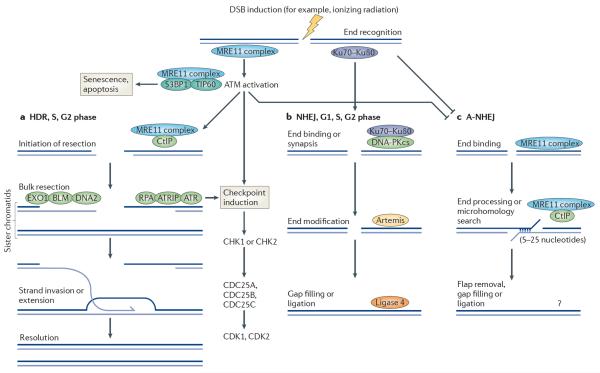
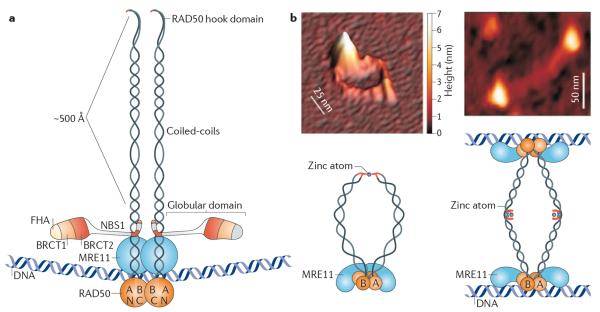
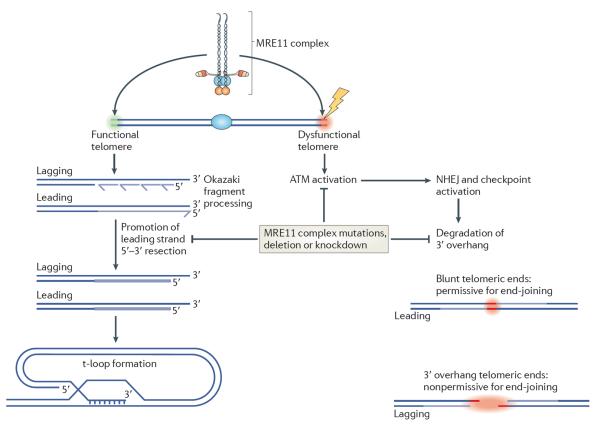
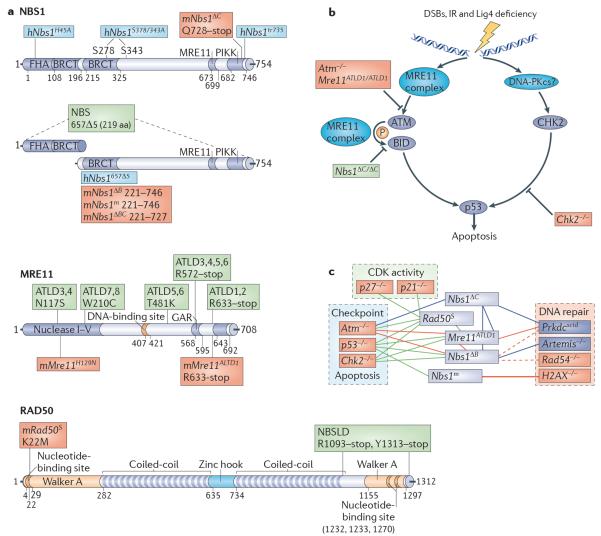
References
-
- Bartkova J, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. - PubMed
-
- Gorgoulis VG, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. - PubMed
-
- Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst.) 2004;3:845–854. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
