

Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy
- PMID: 21257612
- PMCID: PMC3115280
- DOI: 10.1093/cvr/cvr015
Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy
Abstract
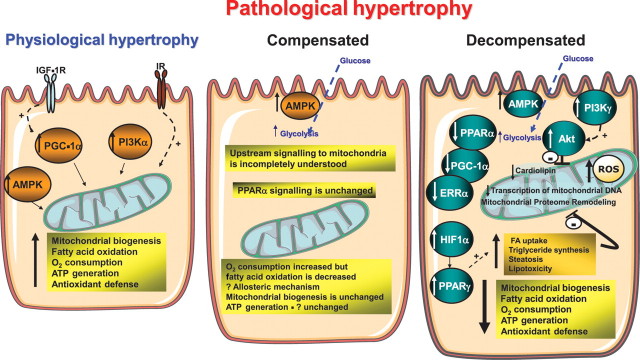
Cardiac hypertrophy is a stereotypic response of the heart to increased workload. The nature of the workload increase may vary depending on the stimulus (repetitive, chronic, pressure, or volume overload). If the heart fully adapts to the new loading condition, the hypertrophic response is considered physiological. If the hypertrophic response is associated with the ultimate development of contractile dysfunction and heart failure, the response is considered pathological. Although divergent signalling mechanisms may lead to these distinct patterns of hypertrophy, there is some overlap. Given the close relationship between workload and energy demand, any form of cardiac hypertrophy will impact the energy generation by mitochondria, which are the key organelles for cellular ATP production. Significant changes in the expression of nuclear and mitochondrially encoded transcripts that impact mitochondrial function as well as altered mitochondrial proteome composition and mitochondrial energetics have been described in various forms of cardiac hypertrophy. Here, we review mitochondrial alterations in pathological and physiological hypertrophy. We suggest that mitochondrial adaptations to pathological and physiological hypertrophy are distinct, and we shall review potential mechanisms that might account for these differences.
Figures
References
-
- Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56:56–64. doi:10.1172/JCI108079. - DOI - PMC - PubMed
-
- Papademetriou V. From hypertension to heart failure. J Clin hypertens (Greenwich) 2004;6:14–17. doi:10.1111/j.1524-6175.2004.03919.x. - DOI - PMC - PubMed
-
- Liu SK, Magid NR, Fox PR, Goldfine SM, Borer JS. Fibrosis, myocyte degeneration and heart failure in chronic experimental aortic regurgitation. Cardiology. 1998;90:101–109. doi:10.1159/000006827. - DOI - PubMed
-
- Bristow MR. Mechanisms of development of heart failure in the hypertensive patient. Cardiology. 1999;92(Suppl. 1)):3–6. discussion 7–9, 20–21 doi:10.1159/000047287. - DOI - PubMed
-
- Carabello BA. Aortic stenosis: from pressure overload to heart failure. Heart Fail Clin. 2006;2:435–442. doi:10.1016/j.hfc.2006.11.001. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
