

Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: a mechanism integrating glycolytic flux with lipid metabolism
- PMID: 21258134
- PMCID: PMC3069396
- DOI: 10.1074/jbc.M110.203661
Reversible high affinity inhibition of phosphofructokinase-1 by acyl-CoA: a mechanism integrating glycolytic flux with lipid metabolism
Abstract
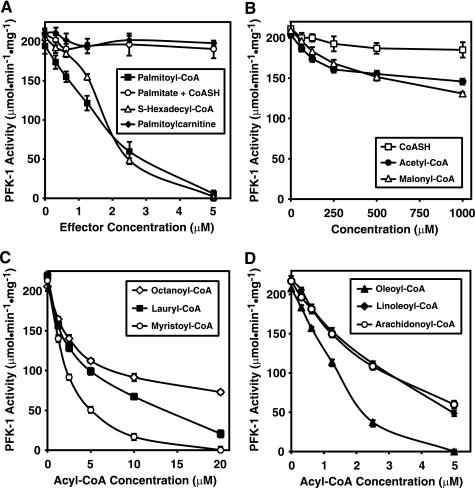
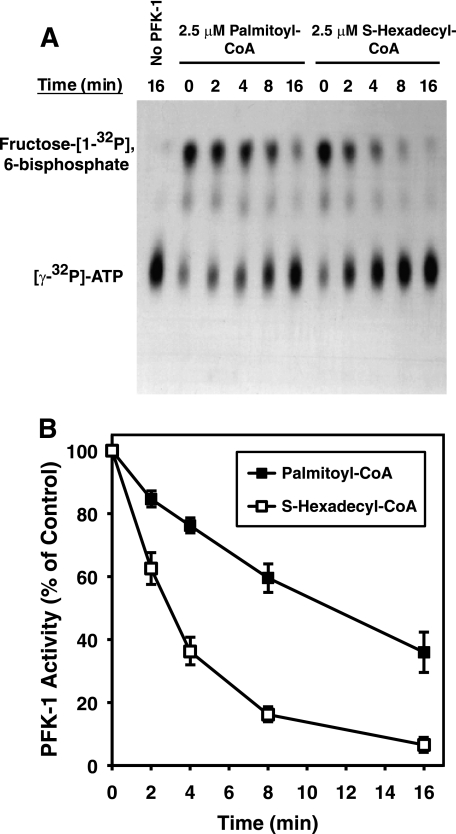
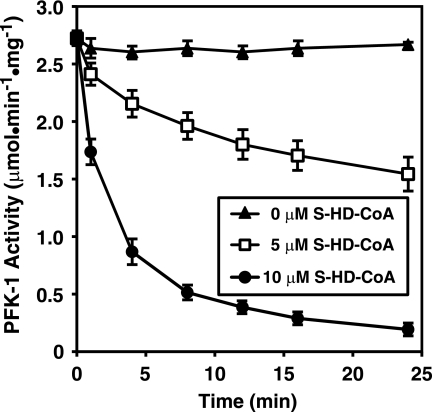
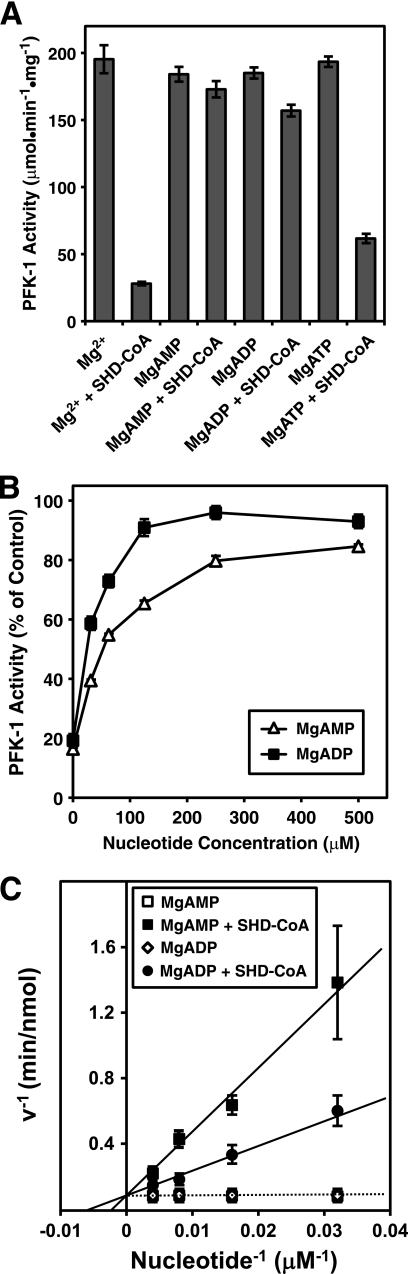
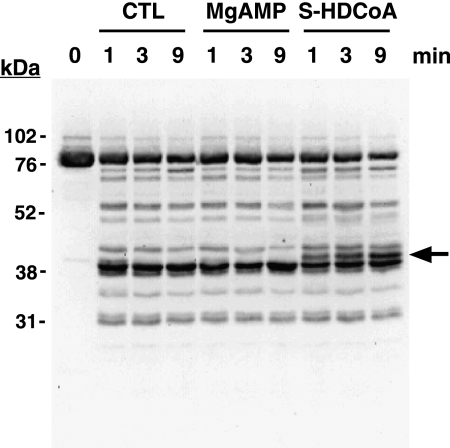
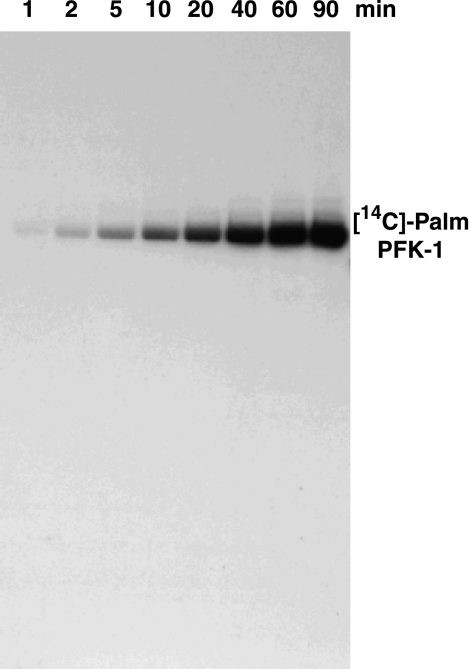
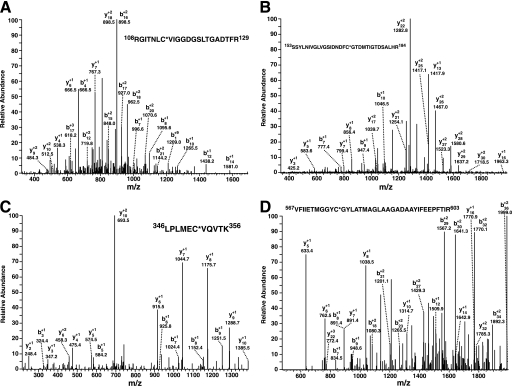
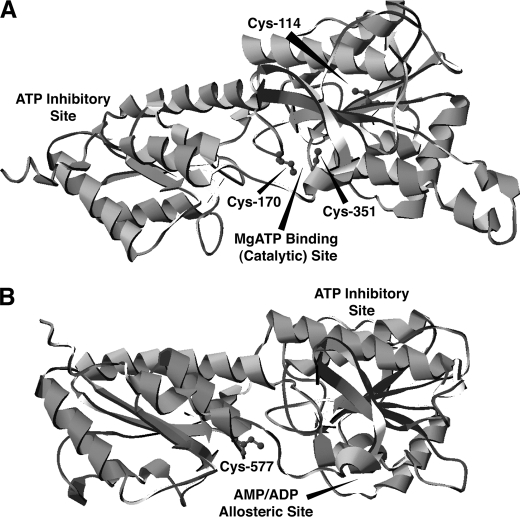
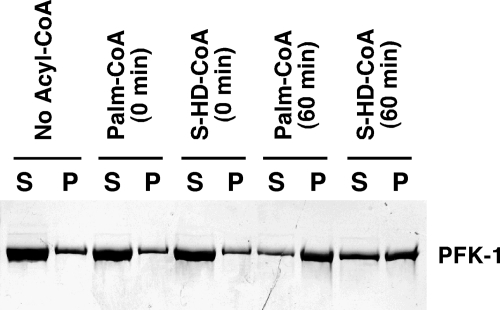
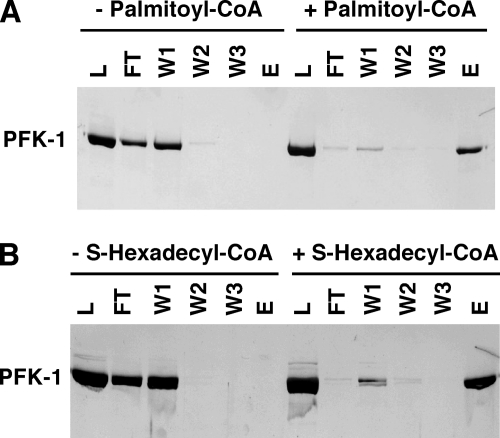
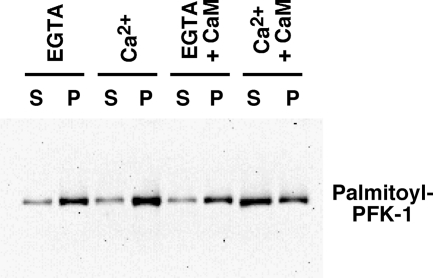
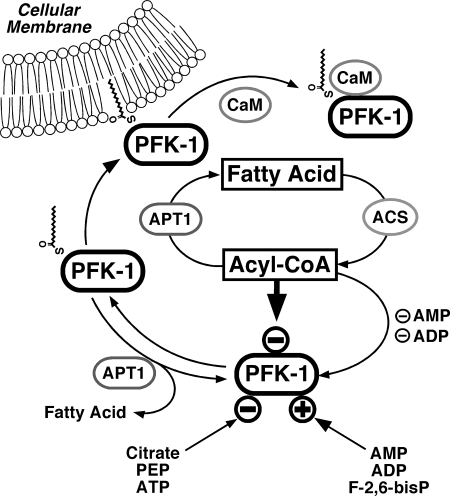
The enzyme phosphofructokinase-1 (PFK-1) catalyzes the first committed step of glycolysis and is regulated by a complex array of allosteric effectors that integrate glycolytic flux with cellular bioenergetics. Here, we demonstrate the direct, potent, and reversible inhibition of purified rabbit muscle PFK-1 by low micromolar concentrations of long chain fatty acyl-CoAs (apparent Ki∼1 μM). In sharp contrast, short chain acyl-CoAs, palmitoylcarnitine, and palmitic acid in the presence of CoASH were without effect. Remarkably, MgAMP and MgADP but not MgATP protected PFK-1 against inhibition by palmitoyl-CoA indicating that acyl-CoAs regulate PFK-1 activity in concert with cellular high energy phosphate status. Furthermore, incubation of PFK-1 with [1-(14)C]palmitoyl-CoA resulted in robust acylation of the enzyme that was reversible by incubation with acyl-protein thioesterase-1 (APT1). Importantly, APT1 reversed palmitoyl-CoA-mediated inhibition of PFK-1 activity. Mass spectrometric analyses of palmitoylated PFK-1 revealed four sites of acylation, including Cys-114, Cys-170, Cys-351, and Cys-577. PFK-1 in both skeletal muscle extracts and in purified form was inhibited by S-hexadecyl-CoA, a nonhydrolyzable palmitoyl-CoA analog, demonstrating that covalent acylation of PFK-1 was not required for inhibition. Tryptic footprinting suggested that S-hexadecyl-CoA induced a conformational change in PFK-1. Both palmitoyl-CoA and S-hexadecyl-CoA increased the association of PFK-1 with Ca2+/calmodulin, which attenuated the binding of palmitoylated PFK-1 to membrane vesicles. Collectively, these results demonstrate that fatty acyl-CoA modulates phosphofructokinase activity through both covalent and noncovalent interactions to regulate glycolytic flux and enzyme membrane localization via the branch point metabolic node that mediates lipid flux through anabolic and catabolic pathways.
Figures

References
-
- Shipp J. C., Opie L. H., Challoner D. (1961) Nature 189, 1018–1019
-
- Newsholme E. A., Randle P. J., Manchester K. L. (1962) Nature 193, 270–271 - PubMed
-
- Garland P. B., Randle P. J., Newsholme E. A. (1963) Nature 200, 169–170 - PubMed
-
- Randle P. J., Garland P. B., Hales C. N., Newsholme E. A. (1963) Lancet 1, 785–789 - PubMed
-
- Lopaschuk G. D. (2002) Heart Fail. Rev. 7, 149–159 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
