

Understanding the specificity of a docking interaction between JNK1 and the scaffolding protein JIP1
- PMID: 21261310
- PMCID: PMC3042137
- DOI: 10.1021/jp1073522
Understanding the specificity of a docking interaction between JNK1 and the scaffolding protein JIP1
Abstract
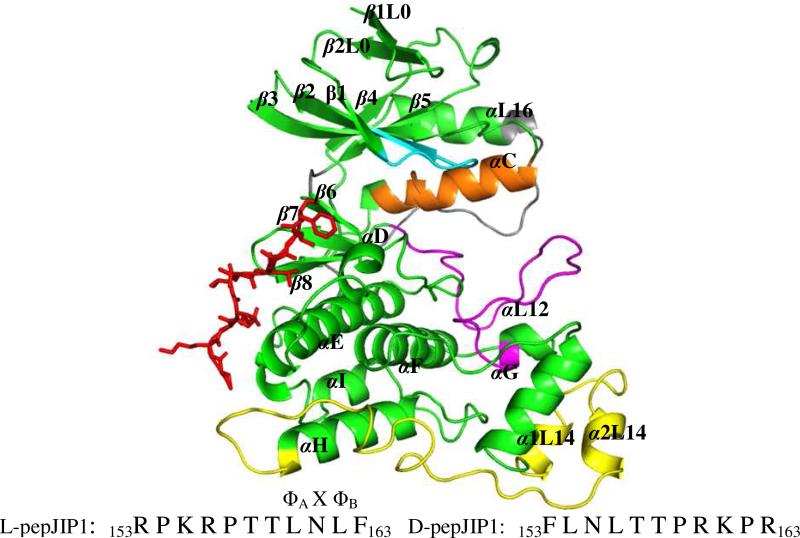
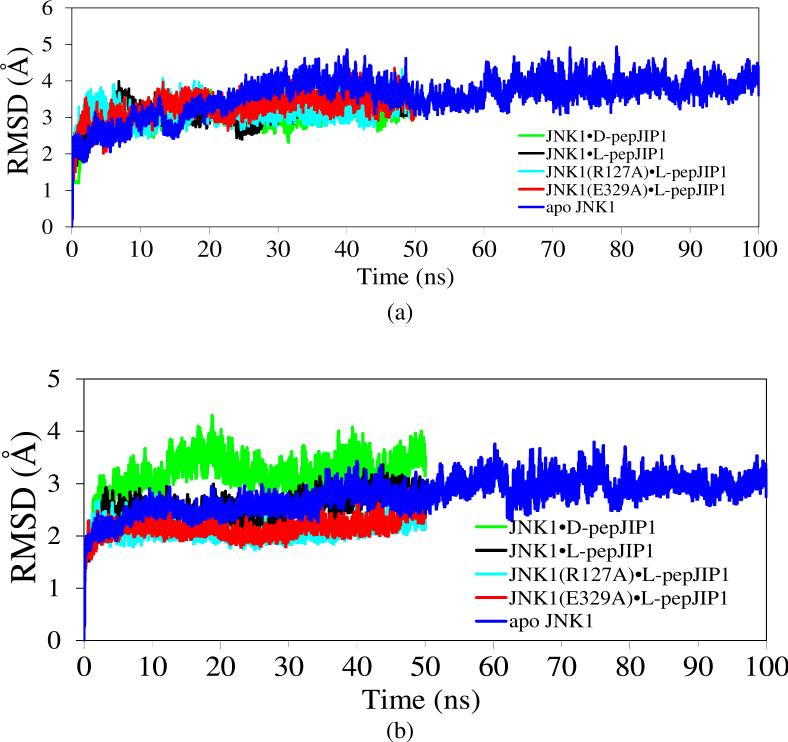
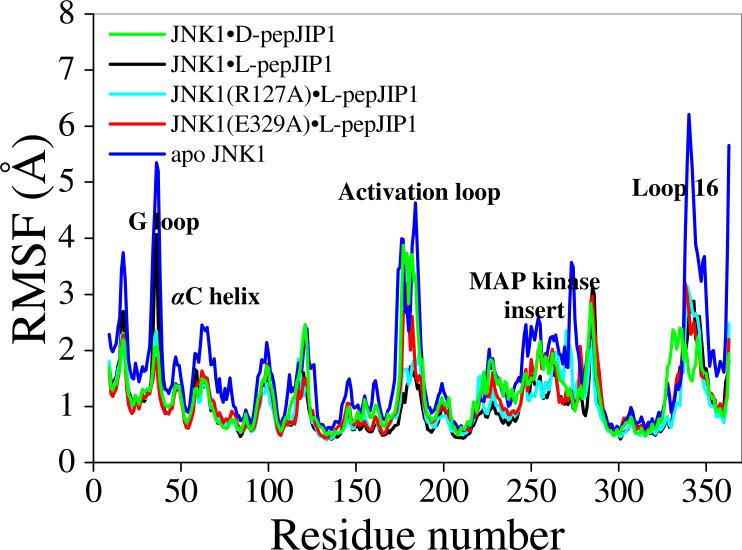
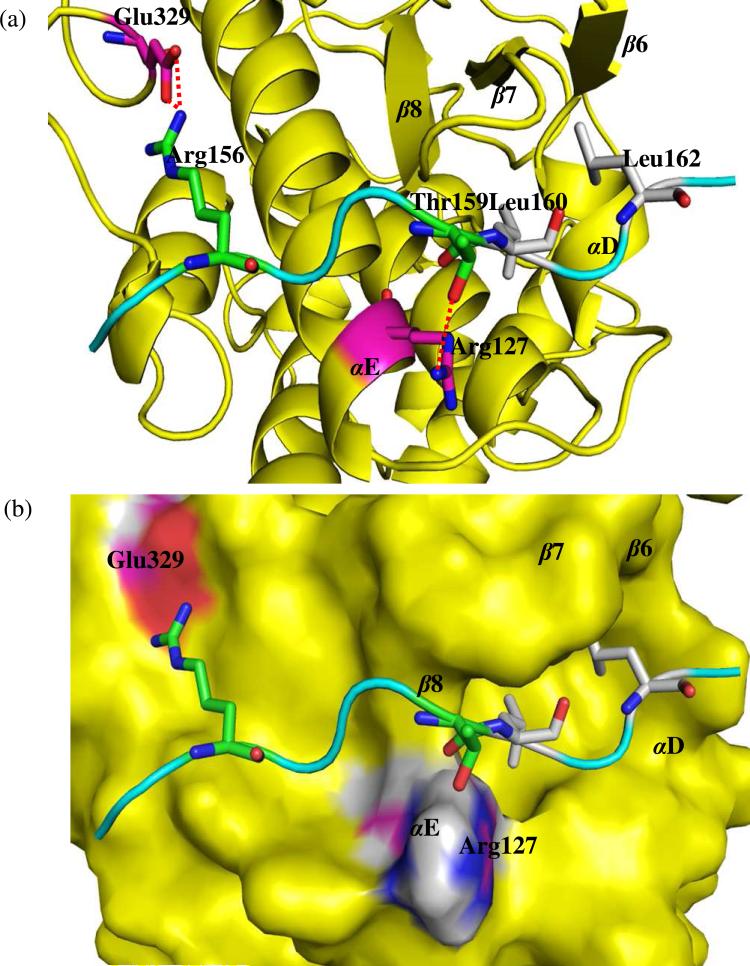
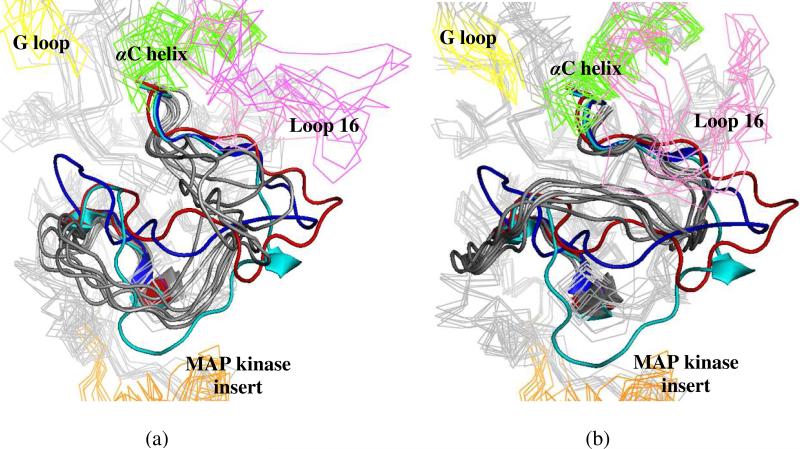
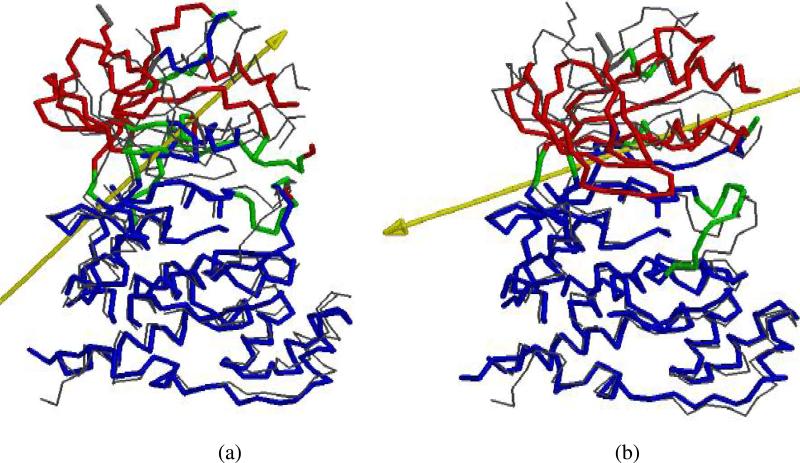
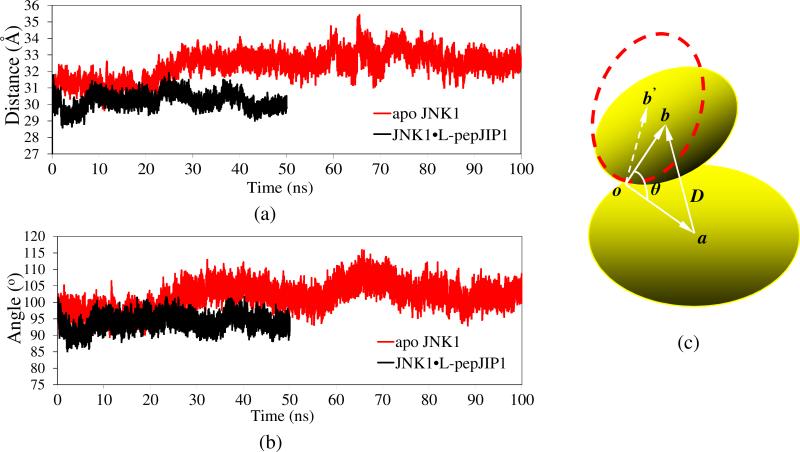
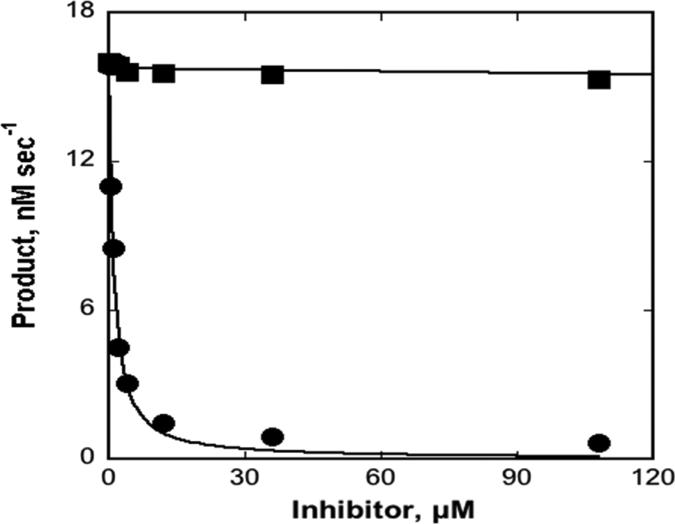
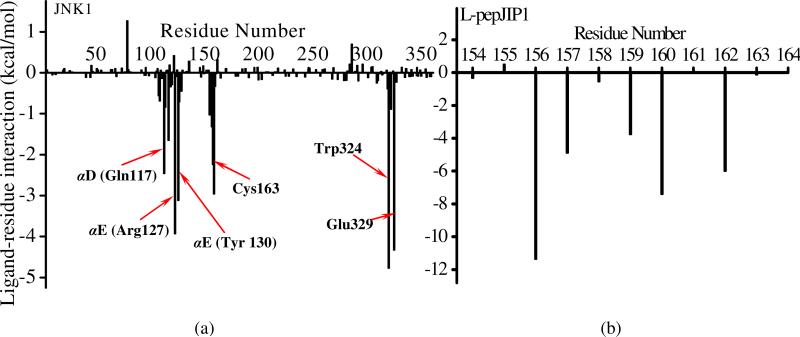
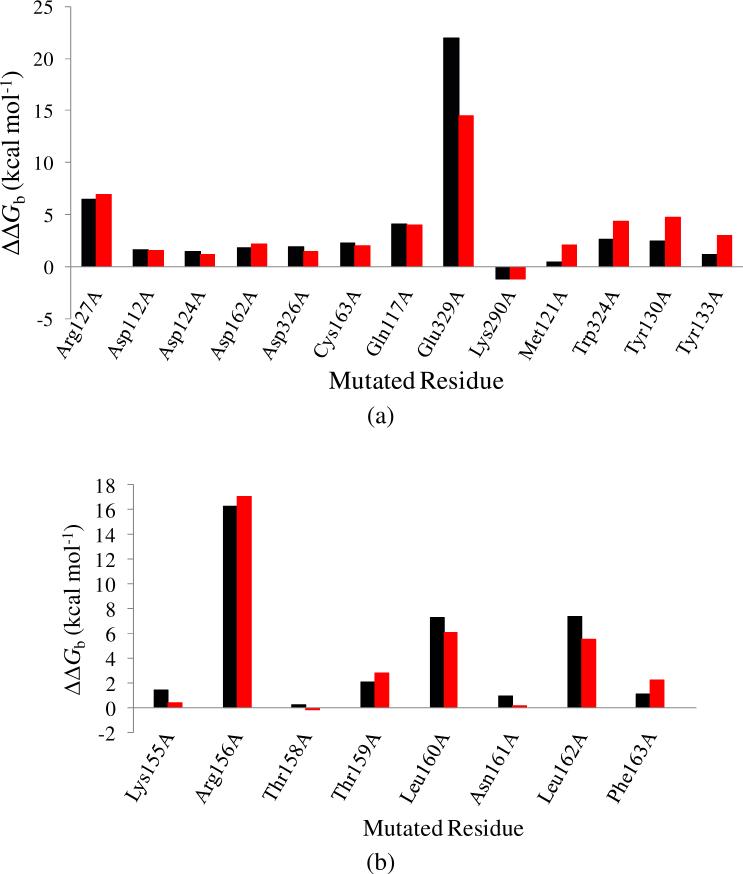
The up-regulation of JNK activity is associated with a number of disease states. The JNK-JIP1 interaction represents an attractive target for the inhibition of JNK-mediated signaling. In this study, molecular dynamics simulations have been performed on the apo-JNK1 and the JNK1•L-pepJIP1 and JNK1•D-pepJIP1 complexes to investigate the interaction between the JIP1 peptides and JNK1. Dynamic domain studies based on essential dynamics (ED) analysis of apo-JNK1 and the JNK1•L-pepJIP1 complex have been performed to analyze and compare details of conformational changes, hinge axes, and hinge bending regions in both structures. The activation loop, the αC helix, and the G loop are found to be highly flexible and to exhibit significant changes in dynamics upon L-pepJIP1 binding. The conformation of the activation loop for the apo state is similar to that of inactive apo-ERK2, while the activation loop in JNK1•L-pepJIP1 complex resembles that of the inactive ERK2 bound with pepHePTP. ED analysis shows that, after the binding of l-pepJIP1, the N- and C-terminal domains of JNK1 display both a closure and a twisting motion centered around the activation loop, which functions as a hinge. In contrast, no domain motion is detected for the apo state for which an open conformation is favored. The present study suggests that L-pepJIP1 regulates the interdomain motions of JNK1 and potentially the active site via an allosteric mechanism. The binding free energies of L-pepJIP1 and D-pepJIP1 to JNK1 are estimated using the molecular mechanics Poisson-Boltzmann and generalized-Born surface area (MM-PB/GBSA) methods. The contribution of each residue at the interaction interface to the binding affinity of L-pepJIP1 with JNK1 has been analyzed by means of computational alanine-scanning mutagenesis and free energy decomposition. Several critical interactions for binding (e.g., Arg156/L-pepJIP1 and Glu329/JNK1) have been identified. The binding free energy calculation indicates that the electrostatic interaction contributes critically to specificity, rather than to binding affinity between the peptide and JNK1. Notably, the binding free energy calculations predict that D-pepJIP1 binding to JNK1 is significantly weaker than the L form, contradicting the previous suggestion that D-pepJIP1 acts as an inhibitor toward JNK1. We have performed experiments using purified JNK1 to confirm that, indeed, D-pepJIP1 does not inhibit the ability of JNK1 to phosphorylate c-Jun in vitro.
Figures

Similar articles
-
Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125.EMBO J. 2004 Jun 2;23(11):2185-95. doi: 10.1038/sj.emboj.7600212. Epub 2004 May 13. EMBO J. 2004. PMID: 15141161 Free PMC article.
-
Bipartite binding of the intrinsically disordered scaffold protein JIP1 to the kinase JNK1.Proc Natl Acad Sci U S A. 2025 Mar 4;122(9):e2419915122. doi: 10.1073/pnas.2419915122. Epub 2025 Feb 25. Proc Natl Acad Sci U S A. 2025. PMID: 39999166 Free PMC article.
-
Understanding the molecular basis of MK2-p38α signaling complex assembly: insights into protein-protein interaction by molecular dynamics and free energy studies.Mol Biosyst. 2012 Aug;8(8):2106-18. doi: 10.1039/c2mb25042j. Epub 2012 May 30. Mol Biosyst. 2012. PMID: 22648002
-
Folding funnels and conformational transitions via hinge-bending motions.Cell Biochem Biophys. 1999;31(2):141-64. doi: 10.1007/BF02738169. Cell Biochem Biophys. 1999. PMID: 10593256 Review.
-
Studying allosteric regulation in metal sensor proteins using computational methods.Adv Protein Chem Struct Biol. 2014;96:181-218. doi: 10.1016/bs.apcsb.2014.06.009. Epub 2014 Sep 6. Adv Protein Chem Struct Biol. 2014. PMID: 25443958 Review.
Cited by
-
Exploring PHD fingers and H3K4me0 interactions with molecular dynamics simulations and binding free energy calculations: AIRE-PHD1, a comparative study.PLoS One. 2012;7(10):e46902. doi: 10.1371/journal.pone.0046902. Epub 2012 Oct 15. PLoS One. 2012. PMID: 23077531 Free PMC article.
-
Arrestin-3-Dependent Activation of c-Jun N-Terminal Kinases (JNKs).Curr Protoc Pharmacol. 2015 Mar 2;68:2.12.1-2.12.26. doi: 10.1002/0471141755.ph0212s68. Curr Protoc Pharmacol. 2015. PMID: 25737158 Free PMC article.
-
Diamidine compounds for selective inhibition of protein arginine methyltransferase 1.J Med Chem. 2014 Mar 27;57(6):2611-22. doi: 10.1021/jm401884z. Epub 2014 Mar 6. J Med Chem. 2014. PMID: 24564570 Free PMC article.
-
Arrestin-3-Dependent Activation of c-Jun N-Terminal Kinases (JNKs).Curr Protoc. 2023 Sep;3(9):e839. doi: 10.1002/cpz1.839. Curr Protoc. 2023. PMID: 37668419 Free PMC article.
-
Development of JNK2-selective peptide inhibitors that inhibit breast cancer cell migration.ACS Chem Biol. 2011 Jun 17;6(6):658-66. doi: 10.1021/cb200017n. Epub 2011 Apr 5. ACS Chem Biol. 2011. PMID: 21438496 Free PMC article.
References
-
- Kyriakis JM, Avruch J. Physiol. Rev. 2001;81:807–869. - PubMed
-
- Pearson G, Robinson F, Gibson TB, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr. Rev. 2001;22:153–183. - PubMed
-
- Derijard B, Hibi M, Wu IH, Barrett T, Su B, Deng TL, Karin M, Davis RJ. Cell. 1994;76:1025–1037. - PubMed
-
- Kallunki T, Su B, Tsigelny I, Sluss HK, Derijard B, Moore G, Davis R, Karin M. Genes Dev. 1994;8:2996–3007. - PubMed
-
- Kyriakis JM, Avruch J. J. Biol. Chem. 1990;265:17355–17363. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
