

The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger
- PMID: 21262234
- PMCID: PMC3094755
- DOI: 10.1016/j.jmb.2011.01.034
The DNA-binding domain of human PARP-1 interacts with DNA single-strand breaks as a monomer through its second zinc finger
Erratum in
- J Mol Biol. 2012 Jun 8;419(3-4):275-6
Abstract
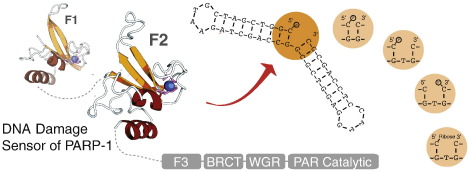
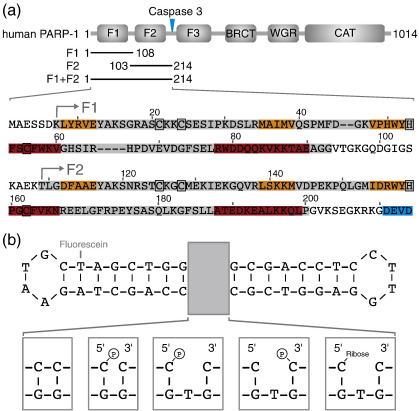
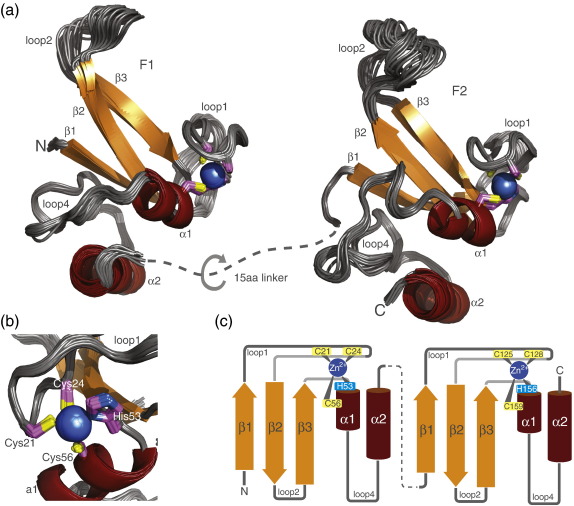
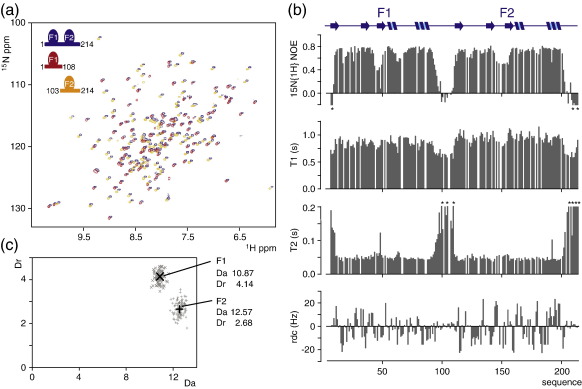
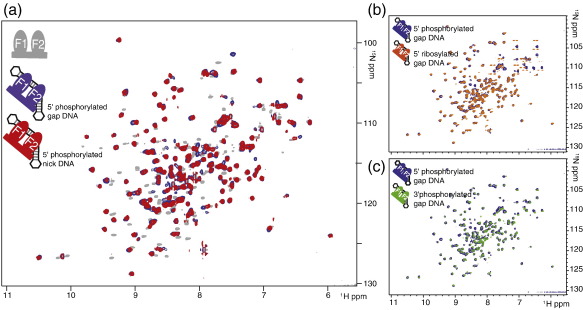
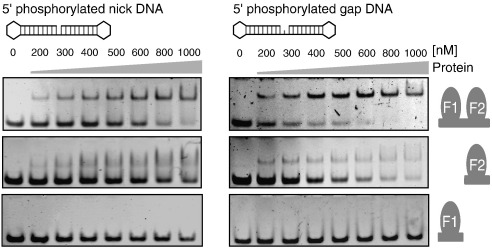
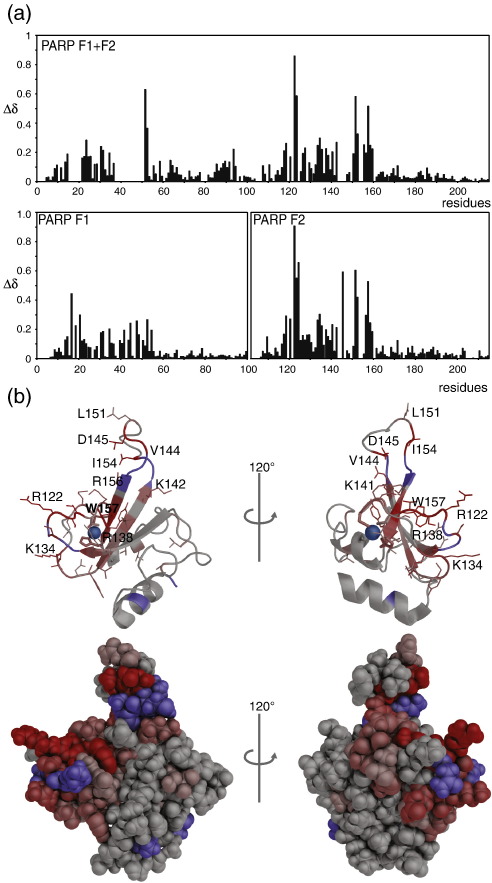
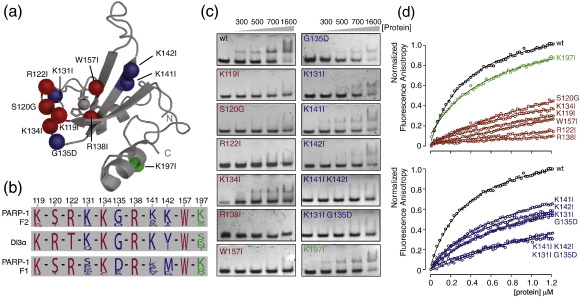
Poly(ADP-ribose)polymerase-1 (PARP-1) is a highly abundant chromatin-associated enzyme present in all higher eukaryotic cell nuclei, where it plays key roles in the maintenance of genomic integrity, chromatin remodeling and transcriptional control. It binds to DNA single- and double-strand breaks through an N-terminal region containing two zinc fingers, F1 and F2, following which its C-terminal catalytic domain becomes activated via an unknown mechanism, causing formation and addition of polyadenosine-ribose (PAR) to acceptor proteins including PARP-1 itself. Here, we report a biophysical and structural characterization of the F1 and F2 fingers of human PARP-1, both as independent fragments and in the context of the 24-kDa DNA-binding domain (F1+F2). We show that the fingers are structurally independent in the absence of DNA and share a highly similar structural fold and dynamics. The F1+F2 fragment recognizes DNA single-strand breaks as a monomer and in a single orientation. Using a combination of NMR spectroscopy and other biophysical techniques, we show that recognition is primarily achieved by F2, which binds the DNA in an essentially identical manner whether present in isolation or in the two-finger fragment. F2 interacts much more strongly with nicked or gapped DNA ligands than does F1, and we present a mutational study that suggests origins of this difference. Our data suggest that different DNA lesions are recognized by the DNA-binding domain of PARP-1 in a highly similar conformation, helping to rationalize how the full-length protein participates in multiple steps of DNA single-strand breakage and base excision repair.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures


References
-
- Ame J.C., Spenlehauer C., de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. - PubMed
-
- Adamietz P. Poly(ADP-ribose) synthase is the major endogenous nonhistone acceptor for poly(ADP-ribose) in alkylated rat hepatoma cells. Eur. J. Biochem. 1987;169:365–372. - PubMed
-
- Tanuma S., Johnson L.D., Johnson G.S. ADP-ribosylation of chromosomal proteins and mouse mammary tumor virus gene expression. Glucocorticoids rapidly decrease endogenous ADP-ribosylation of nonhistone high mobility group 14 and 17 proteins. J. Biol. Chem. 1983;258:15371–15375. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
