

Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence
- PMID: 21266061
- PMCID: PMC3041743
- DOI: 10.1186/1471-2164-12-59
Annotation-based genome-wide SNP discovery in the large and complex Aegilops tauschii genome using next-generation sequencing without a reference genome sequence
Abstract
Background: Many plants have large and complex genomes with an abundance of repeated sequences. Many plants are also polyploid. Both of these attributes typify the genome architecture in the tribe Triticeae, whose members include economically important wheat, rye and barley. Large genome sizes, an abundance of repeated sequences, and polyploidy present challenges to genome-wide SNP discovery using next-generation sequencing (NGS) of total genomic DNA by making alignment and clustering of short reads generated by the NGS platforms difficult, particularly in the absence of a reference genome sequence.
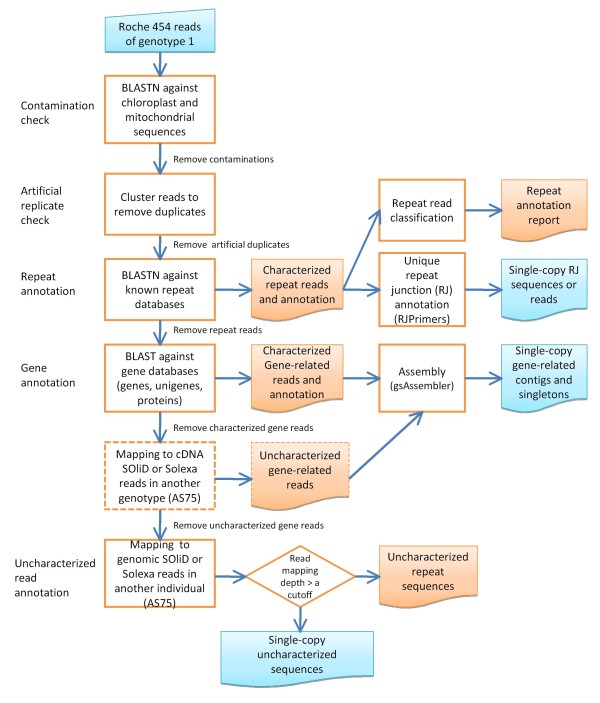
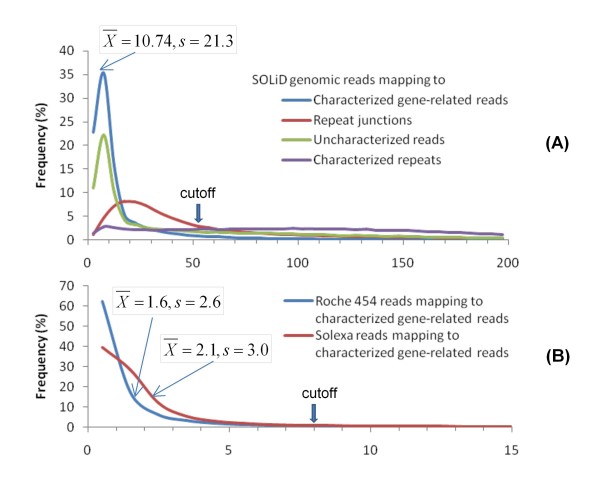
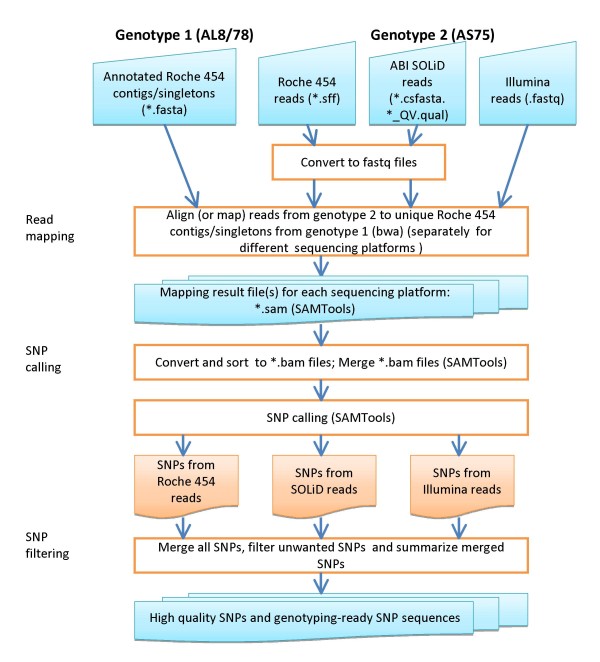
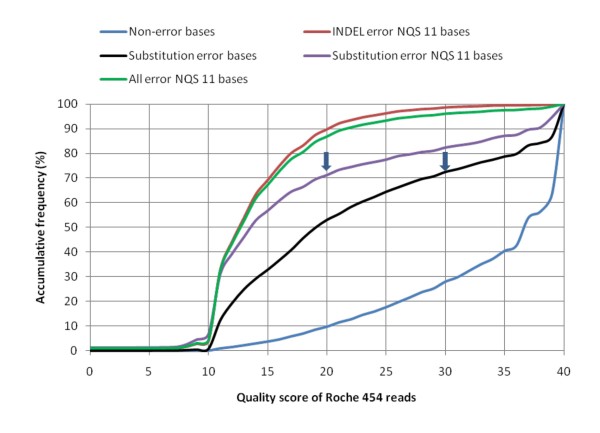
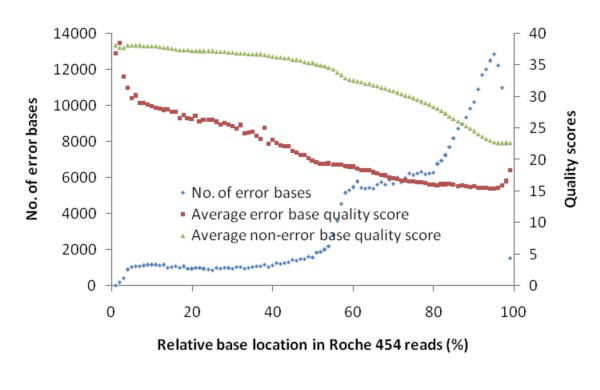
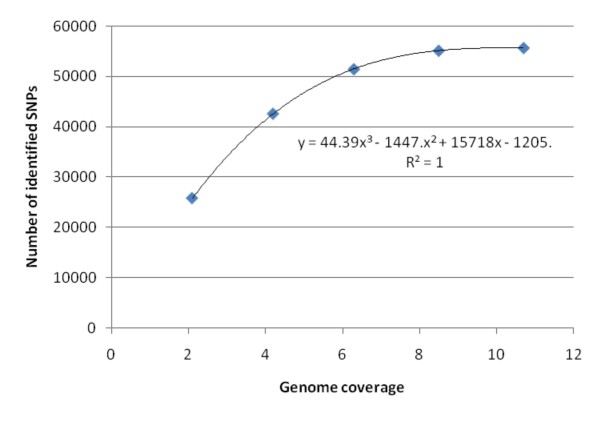
Results: An annotation-based, genome-wide SNP discovery pipeline is reported using NGS data for large and complex genomes without a reference genome sequence. Roche 454 shotgun reads with low genome coverage of one genotype are annotated in order to distinguish single-copy sequences and repeat junctions from repetitive sequences and sequences shared by paralogous genes. Multiple genome equivalents of shotgun reads of another genotype generated with SOLiD or Solexa are then mapped to the annotated Roche 454 reads to identify putative SNPs. A pipeline program package, AGSNP, was developed and used for genome-wide SNP discovery in Aegilops tauschii-the diploid source of the wheat D genome, and with a genome size of 4.02 Gb, of which 90% is repetitive sequences. Genomic DNA of Ae. tauschii accession AL8/78 was sequenced with the Roche 454 NGS platform. Genomic DNA and cDNA of Ae. tauschii accession AS75 was sequenced primarily with SOLiD, although some Solexa and Roche 454 genomic sequences were also generated. A total of 195,631 putative SNPs were discovered in gene sequences, 155,580 putative SNPs were discovered in uncharacterized single-copy regions, and another 145,907 putative SNPs were discovered in repeat junctions. These SNPs were dispersed across the entire Ae. tauschii genome. To assess the false positive SNP discovery rate, DNA containing putative SNPs was amplified by PCR from AL8/78 and AS75 and resequenced with the ABI 3730 xl. In a sample of 302 randomly selected putative SNPs, 84.0% in gene regions, 88.0% in repeat junctions, and 81.3% in uncharacterized regions were validated.
Conclusion: An annotation-based genome-wide SNP discovery pipeline for NGS platforms was developed. The pipeline is suitable for SNP discovery in genomic libraries of complex genomes and does not require a reference genome sequence. The pipeline is applicable to all current NGS platforms, provided that at least one such platform generates relatively long reads. The pipeline package, AGSNP, and the discovered 497,118 Ae. tauschii SNPs can be accessed at (http://avena.pw.usda.gov/wheatD/agsnp.shtml).
Figures
References
-
- Van Tassell CP, Smith TP, Matukumalli LK, Taylor JF, Schnabel RD, Lawley CT, Haudenschild CD, Moore SS, Warren WC, Sonstegard TS. SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries. Nat Methods. 2008;5(3):247–252. doi: 10.1038/nmeth.1185. - DOI - PubMed
-
- Deschamps S, Rota ML, Ratashak JP, Biddle P, Thureen D, Farmer A, Luck S, Beatty M, Nagasawa N, Michael L. et al. Rapid genome-wide single nucleotide polymorphism discovery in soybean and rice via deep resequencing of reduced representation libraries with the Illumina genome analyzer. The Plant Genome. 2010;3(1):53–68. doi: 10.3835/plantgenome2009.09.0026. - DOI
-
- Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC, Specht JE, Farmer AD, May GD, Cregan PB. High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics. 2010;11:38. doi: 10.1186/1471-2164-11-38. - DOI - PMC - PubMed
