

Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis
- PMID: 21266987
- PMCID: PMC3105300
- DOI: 10.1038/ncomms1137
Systems-wide temporal proteomic profiling in glucose-starved Bacillus subtilis
Abstract
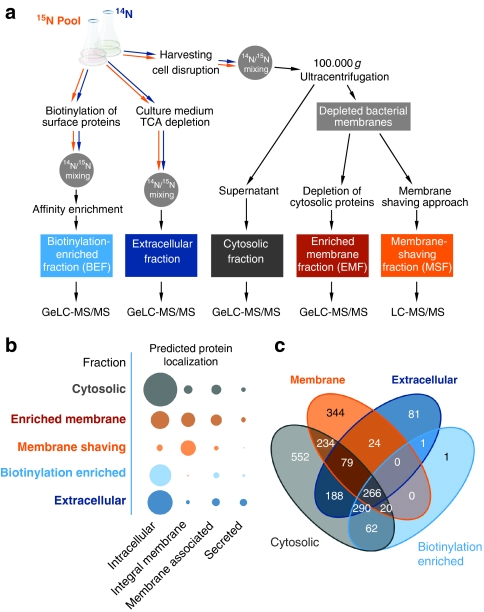
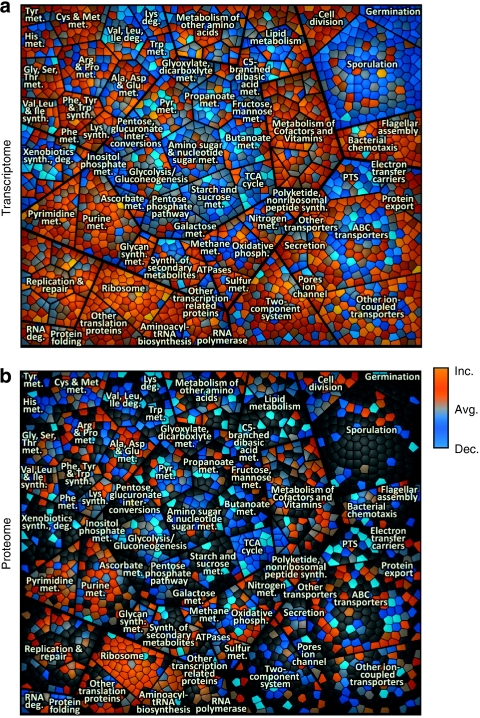
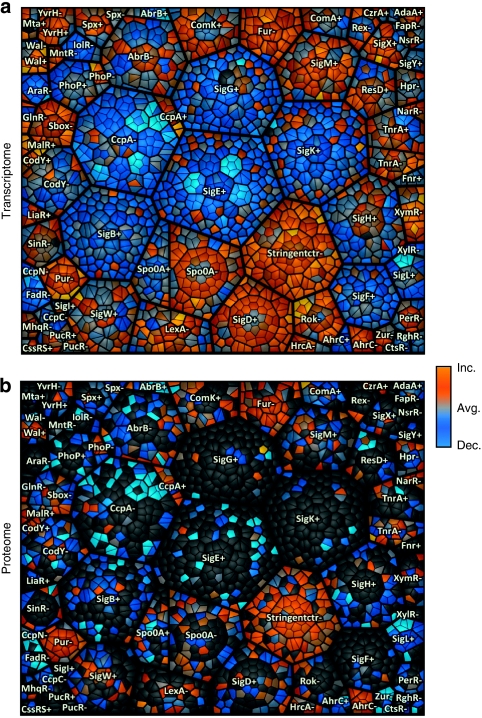
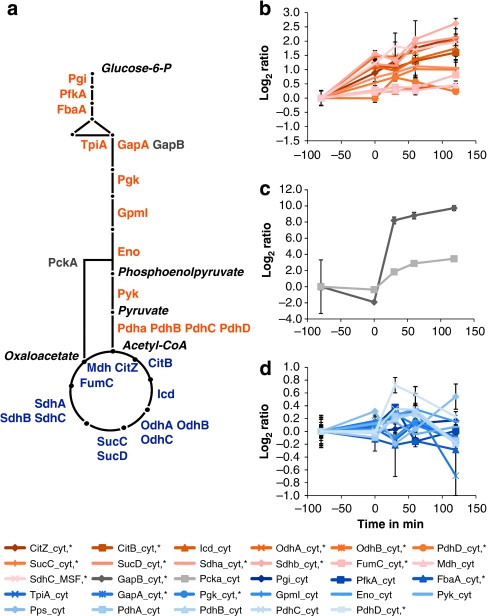
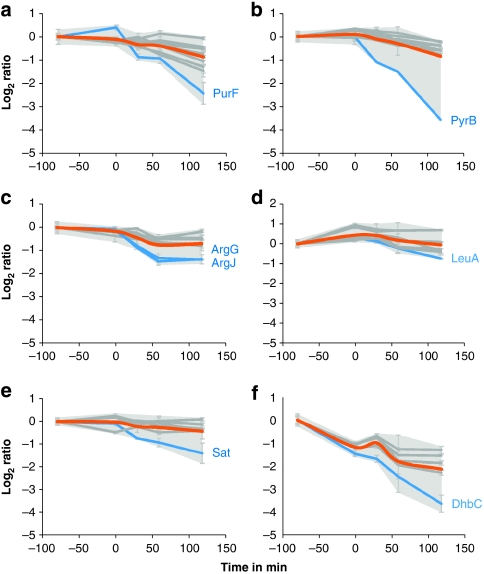
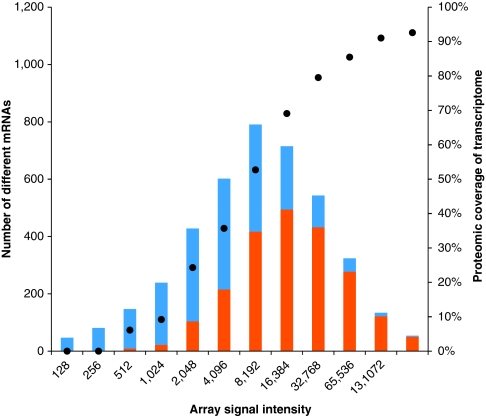
Functional genomics of the Gram-positive model organism Bacillus subtilis reveals valuable insights into basic concepts of cell physiology. In this study, we monitor temporal changes in the proteome, transcriptome and extracellular metabolome of B. subtilis caused by glucose starvation. For proteomic profiling, a combination of in vivo metabolic labelling and shotgun mass spectrometric analysis was carried out for five different proteomic subfractions (cytosolic, integral membrane, membrane, surface and extracellular proteome fraction), leading to the identification of ~52% of the predicted proteome of B. subtilis. Quantitative proteomic and corresponding transcriptomic data were analysed with Voronoi treemaps linking functional classification and relative expression changes of gene products according to their fate in the stationary phase. The obtained data comprise the first comprehensive profiling of changes in the membrane subfraction and allow in-depth analysis of major physiological processes, including monitoring of protein degradation.
Figures
References
-
- Sonenshein A. L. & Hoch J.A.R.L. (eds). Bacillus subtilis and Its Closest Relatives: From Genes to Cells (American Society for Microbiology Press, 2002).
-
- Koburger T., Weibezahn J., Bernhardt J., Homuth G. & Hecker M. Genome-wide mRNA profiling in glucose starved Bacillus subtilis cells. Mol. Genet. Genomics 274, 1–12 (2005). - PubMed
-
- Eymann C. et al. A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics 4, 2849–2876 (2004). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
