

Defects in beta cell Ca²+ signalling, glucose metabolism and insulin secretion in a murine model of K(ATP) channel-induced neonatal diabetes mellitus
- PMID: 21271337
- PMCID: PMC3245714
- DOI: 10.1007/s00125-010-2039-7
Defects in beta cell Ca²+ signalling, glucose metabolism and insulin secretion in a murine model of K(ATP) channel-induced neonatal diabetes mellitus
Abstract
Aims/hypothesis: Mutations that render ATP-sensitive potassium (K(ATP)) channels insensitive to ATP inhibition cause neonatal diabetes mellitus. In mice, these mutations cause insulin secretion to be lost initially and, as the disease progresses, beta cell mass and insulin content also disappear. We investigated whether defects in calcium signalling alone are sufficient to explain short-term and long-term islet dysfunction.
Methods: We examined the metabolic, electrical and insulin secretion response in islets from mice that become diabetic after induction of ATP-insensitive Kir6.2 expression. To separate direct effects of K(ATP) overactivity on beta cell function from indirect effects of prolonged hyperglycaemia, normal glycaemia was maintained by protective exogenous islet transplantation.
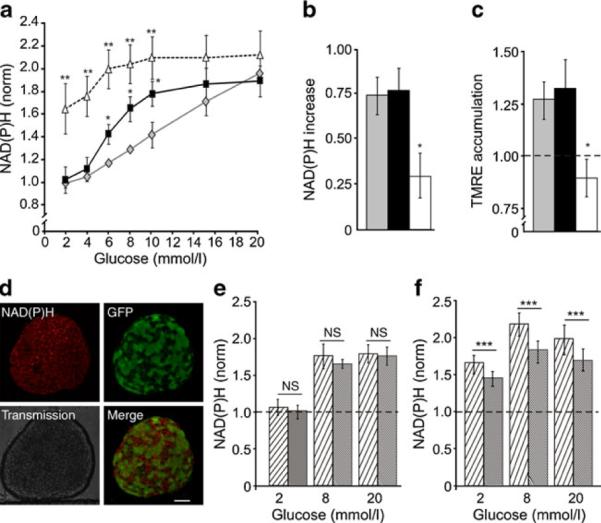
Results: In endogenous islets from protected animals, glucose-dependent elevations of intracellular free-calcium activity ([Ca(2+)](i)) were severely blunted. Insulin content of these islets was normal, and sulfonylureas and KCl stimulated increased [Ca(2+)](i). In the absence of transplant protection, [Ca(2+)](i) responses were similar, but glucose metabolism and redox state were dramatically altered; sulfonylurea- and KCl-stimulated insulin secretion was also lost, because of systemic effects induced by long-term hyperglycaemia and/or hypoinsulinaemia. In both cases, [Ca(2+)](i) dynamics were synchronous across the islet. After reduction of gap-junction coupling, glucose-dependent [Ca(2+)](i) and insulin secretion was partially restored, indicating that excitability of weakly expressing cells is suppressed by cells expressing mutants, via gap-junctions.
Conclusions/interpretation: The primary defect in K(ATP)-induced neonatal diabetes mellitus is failure of glucose metabolism to elevate [Ca(2+)](i), which suppresses insulin secretion and mildly alters islet glucose metabolism. Loss of insulin content and mitochondrial dysfunction are secondary to the long-term hyperglycaemia and/or hypoinsulinaemia that result from the absence of glucose-dependent insulin secretion.
Figures




References
-
- Nichols CG. K-ATP channels as molecular sensors of cellular metabolism. Nature. 2006;440:470–476. - PubMed
-
- Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes—new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54:2503–2513. - PubMed
-
- Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350:1838–1849. - PubMed
-
- Koster JC, Marshall BA, Ensor N, Corbett JA, Nichols CG. Targeted overactivity of beta cell K-ATP channels induces profound neonatal diabetes. Cell. 2000;100:645–654. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
