

Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue
- PMID: 21281512
- PMCID: PMC3038880
- DOI: 10.1186/1756-8935-4-1
Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue
Abstract
Background: Genes subject to genomic imprinting are mono-allelically expressed in a parent-of-origin dependent manner. Each imprinted locus has at least one differentially methylated region (DMR) which has allele specific DNA methylation and contributes to imprinted gene expression. Once DMRs are established, they are potentially able to withstand normal genome reprogramming events that occur during cell differentiation and germ-line DMRs are stably maintained throughout development. These DMRs, in addition to being either maternally or paternally methylated, have differences in whether methylation was acquired in the germ-line or post fertilization and are present in a variety of genomic locations with different Cytosine-phosphate guanine (CpG) densities and CTCF binding capacities. We therefore examined the stability of maintenance of DNA methylation imprints and determined the normal baseline DNA methylation levels in several adult tissues for all imprinted genes. In order to do this, we first developed and validated 50 highly specific, quantitative DNA methylation pyrosequencing assays for the known DMRs associated with human imprinted genes.
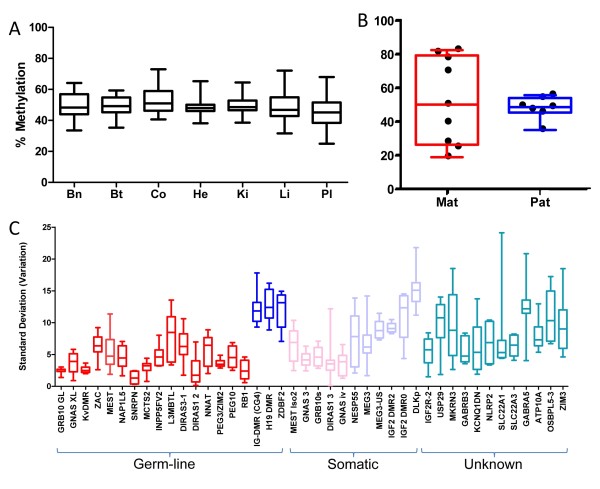
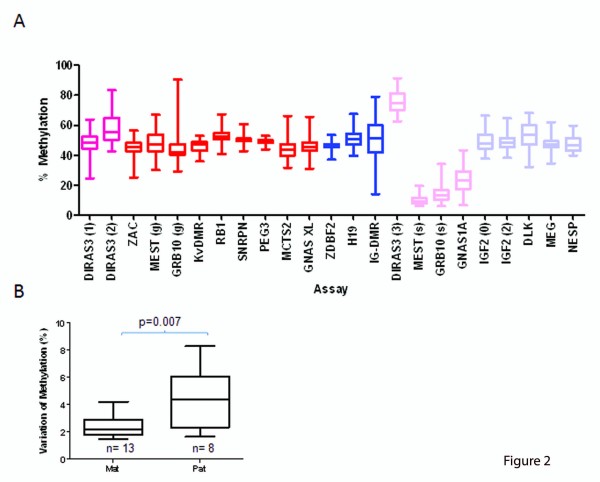
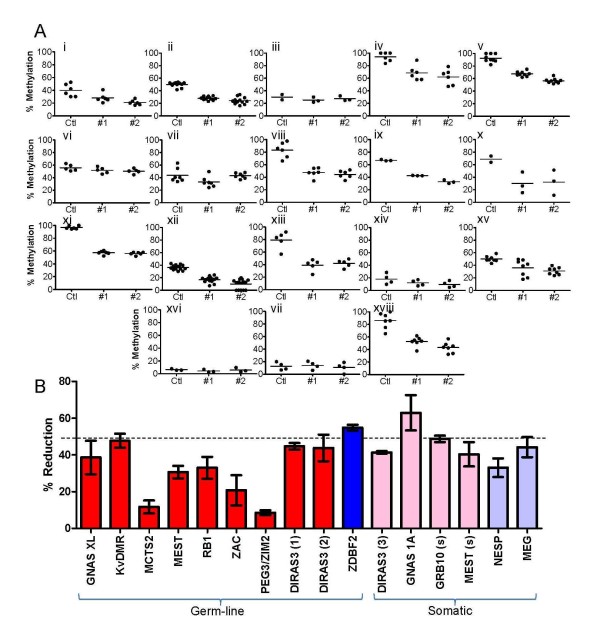
Results: Remarkable stability of the DNA methylation imprint was observed in all germ-line DMRs and paternally methylated somatic DMRs (which maintained average methylation levels of between 35% - 65% in all somatic tissues, independent of gene expression). Maternally methylated somatic DMRs were found to have more variation with tissue specific methylation patterns. Most DMRs, however, showed some intra-individual variability for DNA methylation levels in peripheral blood, suggesting that more than one DMR needs to be examined in order to get an overall impression of the epigenetic stability in a tissue. The plasticity of DNA methylation at imprinted genes was examined in a panel of normal and cancer cell lines. All cell lines showed changes in DNA methylation, especially at the paternal germ-line and the somatic DMRs.
Conclusions: Our validated pyrosequencing methylation assays can be widely used as a tool to investigate DNA methylation levels of imprinted genes in clinical samples. This first comprehensive analysis of normal methylation levels in adult somatic tissues at human imprinted regions confirm that, despite intra-individual variability and tissue specific expression, imprinted genes faithfully maintain their DNA methylation in healthy adult tissue. DNA methylation levels of a selection of imprinted genes are, therefore, a valuable indicator for epigenetic stability.
Figures
References
-
- Novik KL, Nimmrich I, Genc B, Maier S, Piepenbrock C, Olek A, Beck S. Epigenomics: genome-wide study of methylation phenomena. Curr Issues Mol Biol. 2002;4(4):111–128. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
