

Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes
- PMID: 21282188
- PMCID: PMC3063987
- DOI: 10.1093/hmg/ddr037
Mutations in PPIB (cyclophilin B) delay type I procollagen chain association and result in perinatal lethal to moderate osteogenesis imperfecta phenotypes
Abstract
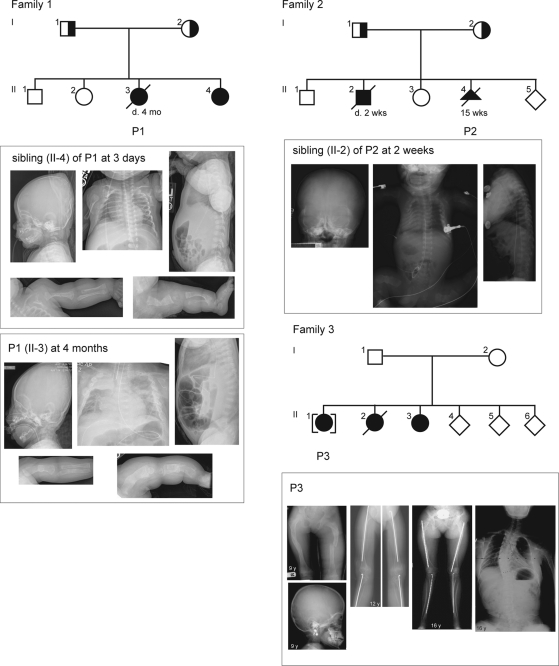
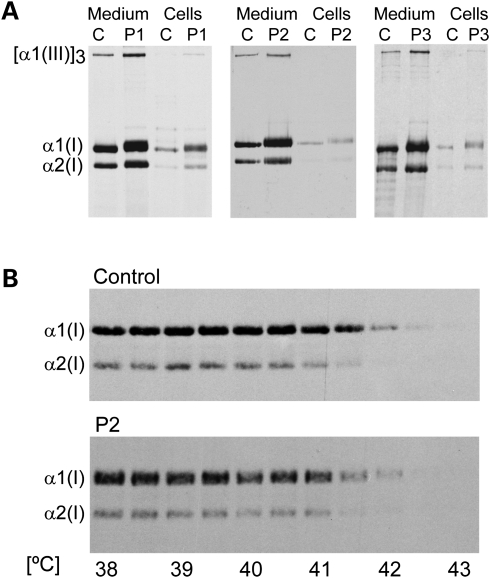
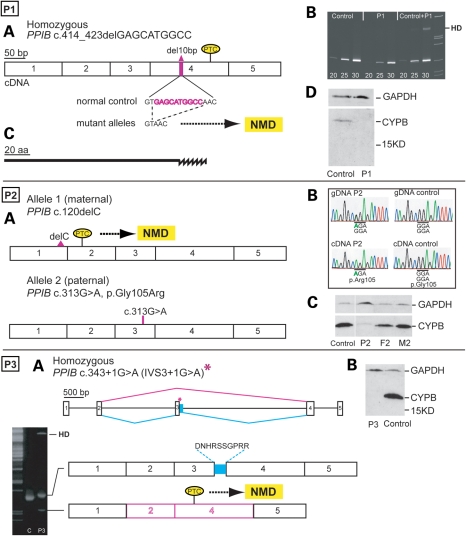
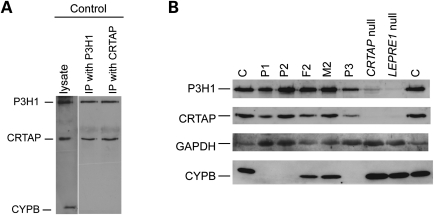
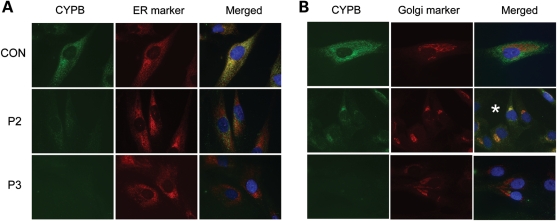
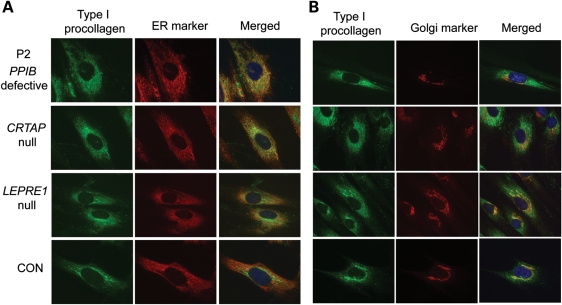
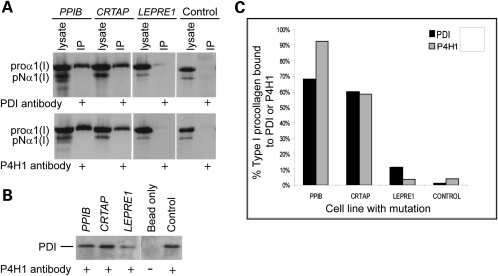
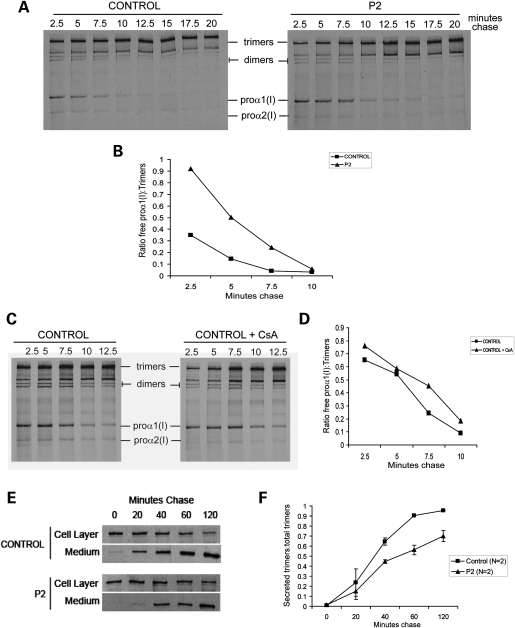
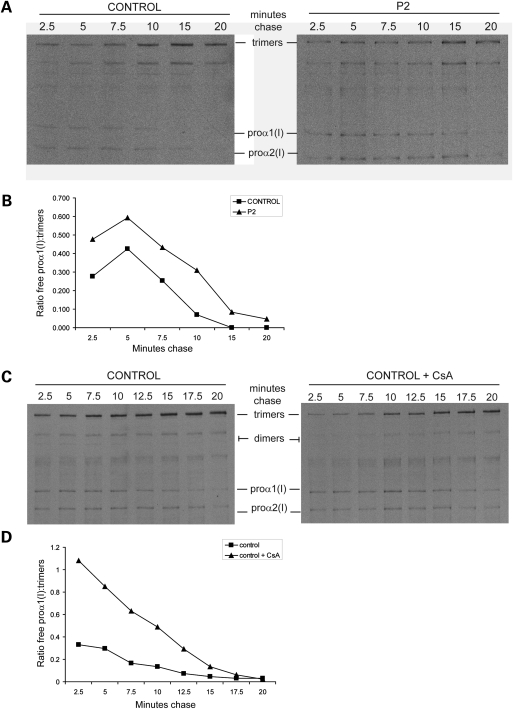
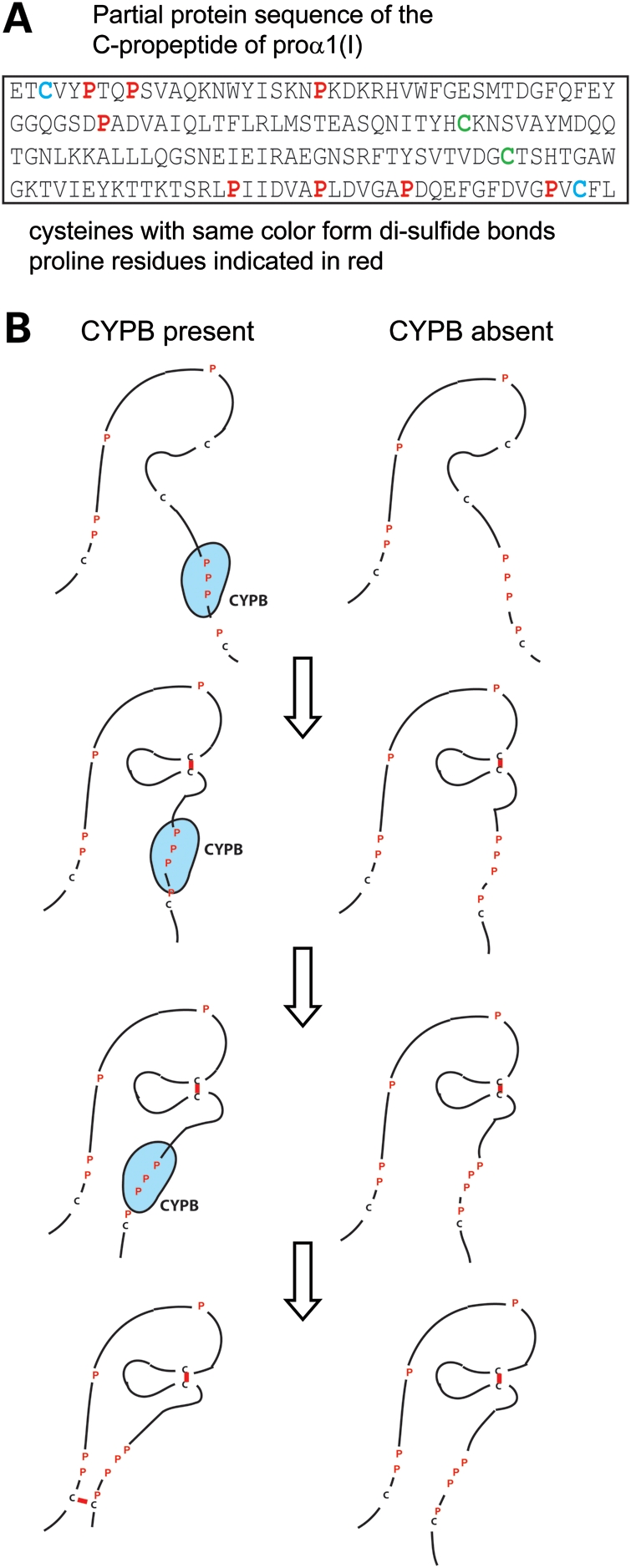
Recessive mutations in the cartilage-associated protein (CRTAP), leucine proline-enriched proteoglycan 1 (LEPRE1) and peptidyl prolyl cis-trans isomerase B (PPIB) genes result in phenotypes that range from lethal in the perinatal period to severe deforming osteogenesis imperfecta (OI). These genes encode CRTAP (encoded by CRTAP), prolyl 3-hydroxylase 1 (P3H1; encoded by LEPRE1) and cyclophilin B (CYPB; encoded by PPIB), which reside in the rough endoplasmic reticulum (RER) and can form a complex involved in prolyl 3-hydroxylation in type I procollagen. CYPB, a prolyl cis-trans isomerase, has been thought to drive the prolyl-containing peptide bonds to the trans configuration needed for triple helix formation. Here, we describe mutations in PPIB identified in cells from three individuals with OI. Cultured dermal fibroblasts from the most severely affected infant make some overmodified type I procollagen molecules. Proα1(I) chains are slow to assemble into trimers, and abnormal procollagen molecules concentrate in the RER, and bind to protein disulfide isomerase (PDI) and prolyl 4-hydroxylase 1 (P4H1). These findings suggest that although CYPB plays a role in helix formation another effect is on folding of the C-terminal propeptide and trimer formation. The extent of procollagen accumulation and PDI/P4H1 binding differs among cells with mutations in PPIB, CRTAP and LEPRE1 with the greatest amount in PPIB-deficient cells and the least in LEPRE1-deficient cells. These findings suggest that prolyl cis-trans isomerase may be required to effectively fold the proline-rich regions of the C-terminal propeptide to allow proα chain association and suggest an order of action for CRTAP, P3H1 and CYPB in procollagen biosynthesis and pathogenesis of OI.
Figures
References
-
- Morello R., Bertin T., Chen Y., Hicks J., Tonachini L., Monticone M., Castagnola P., Rauch F., Glorieux F., Vranka J., et al. CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell. 2006;127:291–304. doi:10.1016/j.cell.2006.08.039. - DOI - PubMed
-
- Barnes A., Chang W., Morello R., Cabral W., Weis M., Eyre D., Leikin S., Makareeva E., Kuznetsova N., Uveges T., et al. Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N. Engl. J. Med. 2006;355:2757–2764. doi:10.1056/NEJMoa063804. - DOI - PMC - PubMed
-
- Cabral W., Chang W., Barnes A., Weis M., Scott M., Leikin S., Makareeva E., Kuznetsova N., Rosenbaum K., Tifft C., et al. Prolyl 3-hydroxylase 1 deficiency causes a recessive metabolic bone disorder resembling lethal/severe osteogenesis imperfecta. Nat. Genet. 2007;39:359–365. doi:10.1038/ng1968. - DOI - PMC - PubMed
-
- Baldridge D., Schwarze U., Morello R., Lennington J., Bertin T., Pace J., Pepin M., Weis M., Eyre D., Walsh J., et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum. Mutat. 2008;29:1435–1442. doi:10.1002/humu.20799. - DOI - PMC - PubMed
-
- Willaert A., Malfait F., Symoens S., Gevaert K., Kayserili H., Megarbane A., Mortier G., Leroy J., Coucke P., De Paepe A. Recessive osteogenesis imperfecta caused by LEPRE1 mutations: clinical documentation and identification of the splice form responsible for prolyl 3-hydroxylation. J. Med. Genet. 2009;46:233–241. doi:10.1136/jmg.2008.062729. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
