

Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer
- PMID: 21283676
- PMCID: PMC3025923
- DOI: 10.1371/journal.pone.0016080
Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer
Abstract
Background: Aberrant DNA methylation patterns might be used as a biomarker for diagnosis and management of cancer patients.
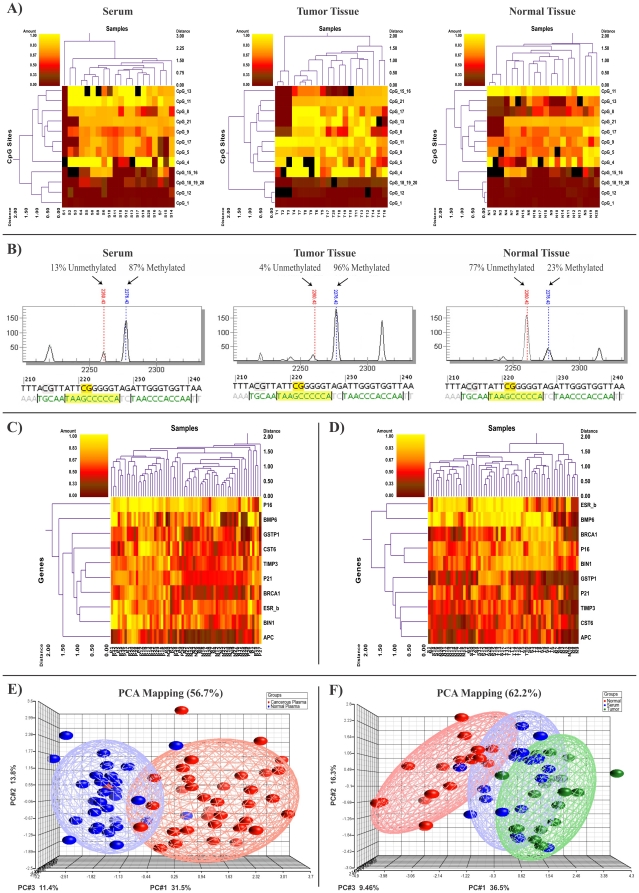
Methods and findings: To achieve a gene panel for developing a breast cancer blood-based test we quantitatively assessed the DNA methylation proportion of 248 CpG sites per sample (total of 31,248 sites in all analyzed samples) on 10 candidate genes (APC, BIN1, BMP6, BRCA1, CST6, ESR-b, GSTP1, P16, P21 and TIMP3). The number of 126 samples consisting of two different cohorts was used (first cohort: plasma samples from breast cancer patients and normal controls; second cohort: triple matched samples including cancerous tissue, matched normal tissue and serum samples). In the first cohort, circulating cell free methylated DNA of the 8 tumor suppressor genes (TSGs) was significantly higher in patients with breast cancer compared to normal controls (P<0.01). In the second cohort containing triple matched samples, seven genes showed concordant hypermethylated profile in tumor tissue and serum samples compared to normal tissue (P<0.05). Using eight genes as a panel to develop a blood-based test for breast cancer, a sensitivity and specificity of more than 90% could be achieved in distinguishing between tumor and normal samples.
Conclusions: Our study suggests that the selected TSG panel combined with the high-throughput technology might be a useful tool to develop epigenetic based predictive and prognostic biomarker for breast cancer relying on pathologic methylation changes in tumor tissue, as well as in circulation.
Conflict of interest statement
Figures



References
-
- Etzioni R, Urban N, Ramsey S, McIntosh M, Schwartz S, et al. The case for early detection. Nat Rev Cancer. 2003;3:243–252. - PubMed
-
- Ries L MD, Krapcho M, Mariotto A, Miller B, Feuer E. SEER cancer statistics review, 1975–2004. Bethesda: National Cancer Institute; 2006.
-
- Radpour R, Barekati Z, Kohler C, Holzgreve W, Zhong XY. New trends in molecular biomarker discovery for breast cancer. Genet Test Mol Biomarkers. 2009;13:565–571. - PubMed
-
- Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646–650. - PubMed
-
- Chan KC, Zhang J, Hui AB, Wong N, Lau TK, et al. Size distributions of maternal and fetal DNA in maternal plasma. Clin Chem. 2004;50:88–92. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
