

Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation
- PMID: 21285515
- PMCID: PMC3049384
- DOI: 10.1172/JCI43584
Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation
Erratum in
-
Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation.J Clin Invest. 2018 Oct 1;128(10):4742-4743. doi: 10.1172/JCI123039. Epub 2018 Oct 1. J Clin Invest. 2018. PMID: 30272582 Free PMC article. No abstract available.
Expression of concern in
-
Particulate allergens potentiate allergic asthma in mice through sustained IgE-mediated mast cell activation.J Clin Invest. 2017 Oct 2;127(10):3913. doi: 10.1172/JCI97321. Epub 2017 Sep 18. J Clin Invest. 2017. PMID: 28920926 Free PMC article. No abstract available.
Abstract
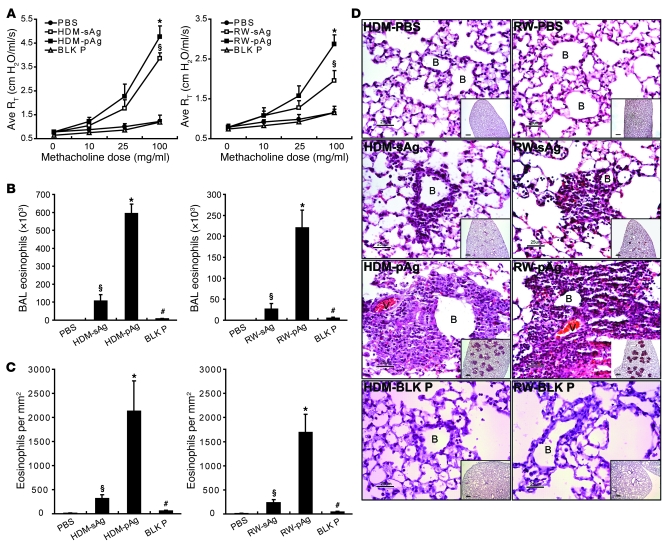
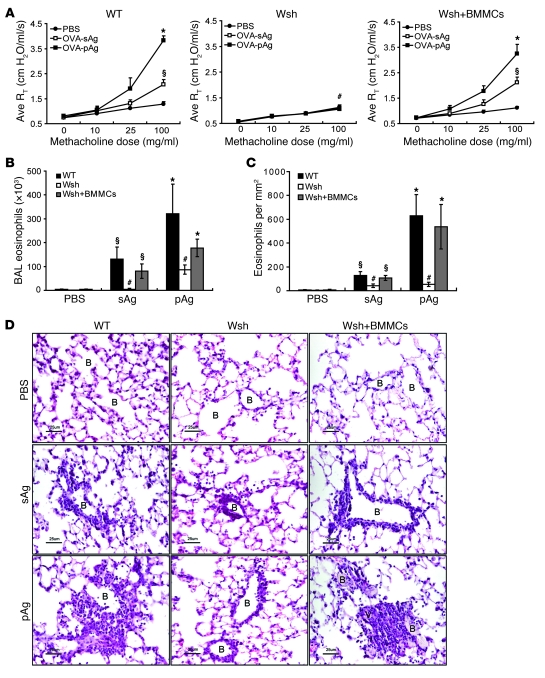
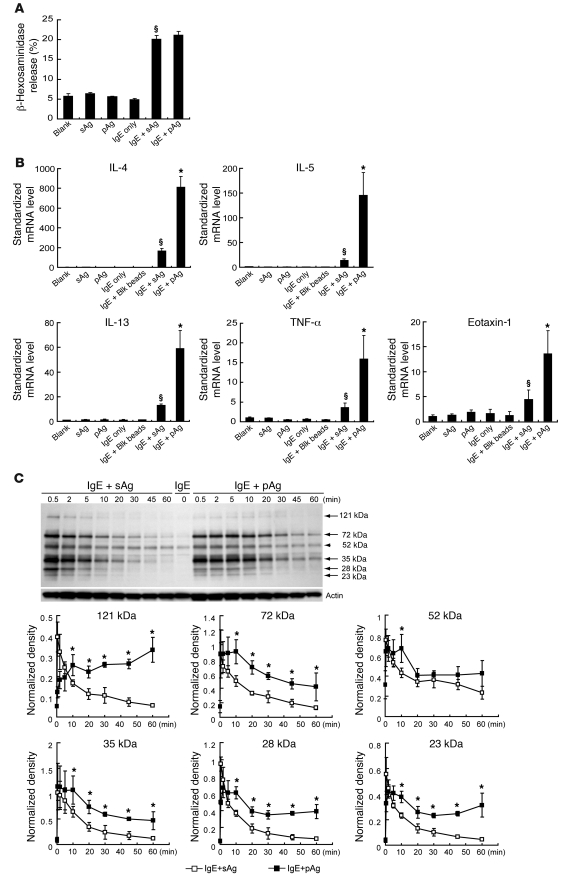
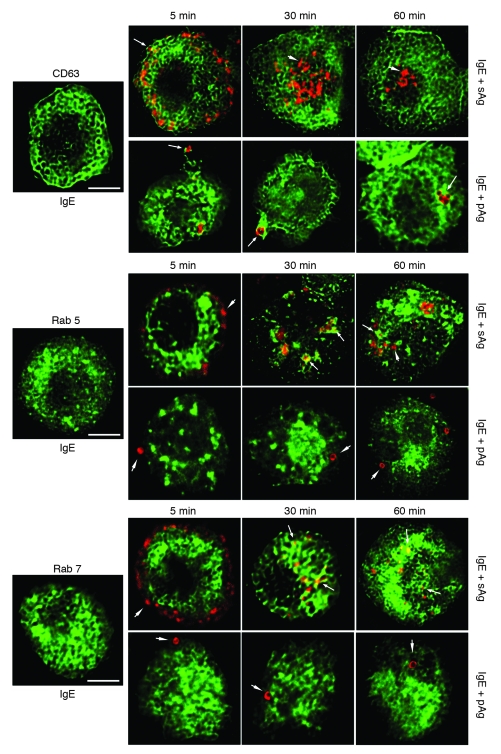
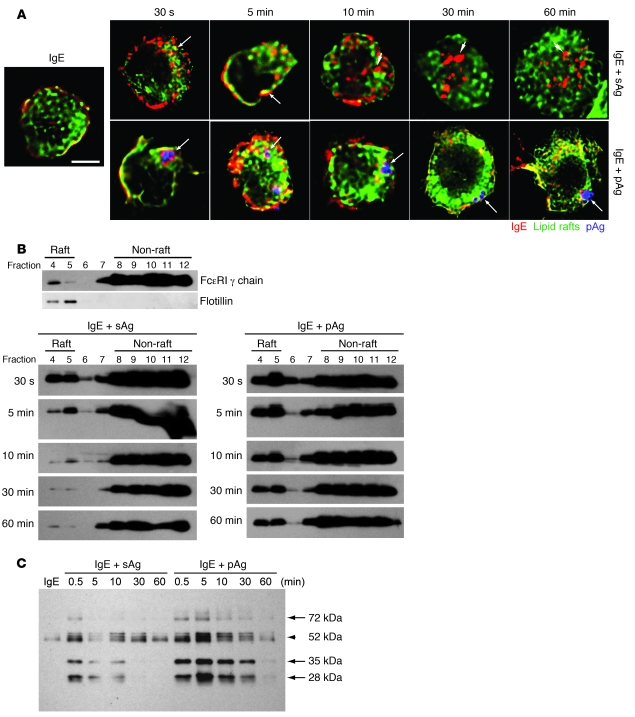
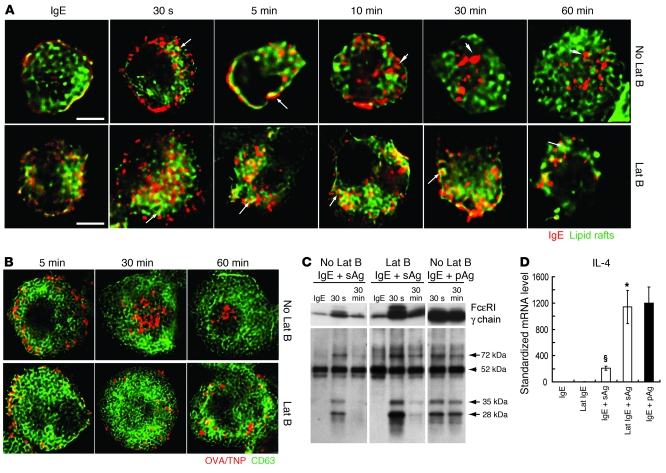
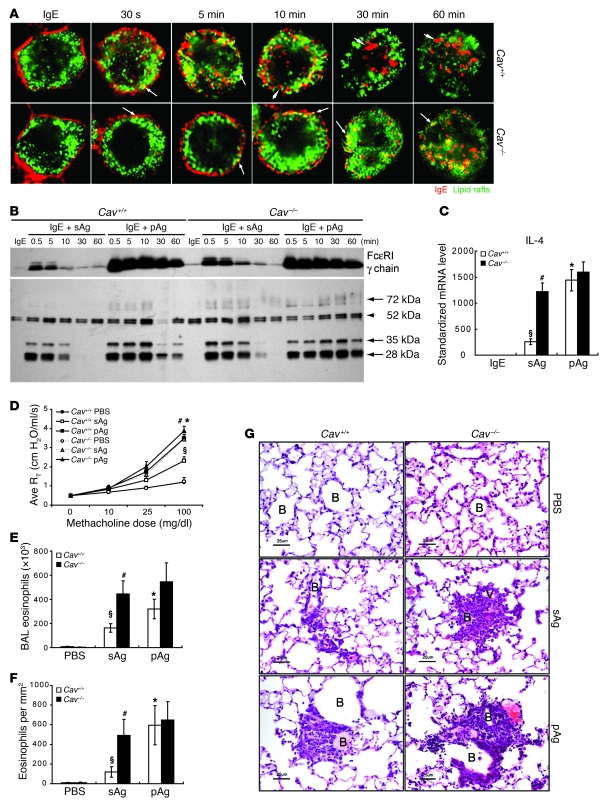
Allergic asthma is characterized by airway hyperresponsiveness, inflammation, and a cellular infiltrate dominated by eosinophils. Numerous epidemiological studies have related the exacerbation of allergic asthma with an increase in ambient inhalable particulate matter from air pollutants. This is because inhalable particles efficiently deliver airborne allergens deep into the airways, where they can aggravate allergic asthma symptoms. However, the cellular mechanisms by which inhalable particulate allergens (pAgs) potentiate asthmatic symptoms remain unknown, in part because most in vivo and in vitro studies exploring the pathogenesis of allergic asthma use soluble allergens (sAgs). Using a mouse model of allergic asthma, we found that, compared with their sAg counterparts, pAgs triggered markedly heightened airway hyperresponsiveness and pulmonary eosinophilia in allergen-sensitized mice. Mast cells (MCs) were implicated in this divergent response, as the differences in airway inflammatory responses provoked by the physical nature of the allergens were attenuated in MC-deficient mice. The pAgs were found to mediate MC-dependent responses by enhancing retention of pAg/IgE/FcεRI complexes within lipid raft–enriched, CD63(+) endocytic compartments, which prolonged IgE/FcεRI-initiated signaling and resulted in heightened cytokine responses. These results reveal how the physical attributes of allergens can co-opt MC endocytic circuitry and signaling responses to aggravate pathological responses of allergic asthma in mice.
Figures
Comment in
-
Findings of Research Misconduct.Fed Regist. 2019 Nov 7;84(216):60097-60098. Fed Regist. 2019. PMID: 37547121 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
- DK077159/DK/NIDDK NIH HHS/United States
- AI074751/AI/NIAID NIH HHS/United States
- R37 DK050814/DK/NIDDK NIH HHS/United States
- P30-AI051445/AI/NIAID NIH HHS/United States
- P30 AI051445/AI/NIAID NIH HHS/United States
- P01 AI081672/AI/NIAID NIH HHS/United States
- R56 AI074751/AI/NIAID NIH HHS/United States
- AI056101/AI/NIAID NIH HHS/United States
- R13 AI150021/AI/NIAID NIH HHS/United States
- ES016347/ES/NIEHS NIH HHS/United States
- U54-AI057157/AI/NIAID NIH HHS/United States
- R01 ES016347/ES/NIEHS NIH HHS/United States
- UC6 AI058607/AI/NIAID NIH HHS/United States
- DK077307/DK/NIDDK NIH HHS/United States
- R01 DK077159/DK/NIDDK NIH HHS/United States
- R21 DK077307/DK/NIDDK NIH HHS/United States
- R21 AI056101/AI/NIAID NIH HHS/United States
- AI081672/AI/NIAID NIH HHS/United States
- UC6-AI058607/AI/NIAID NIH HHS/United States
- DK050814/DK/NIDDK NIH HHS/United States
- U54 AI057157/AI/NIAID NIH HHS/United States
- AI150021/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
