

Review
doi: 10.1172/JCI45691.
Epub 2011 Feb 1.
Energy deficit in Huntington disease: why it matters
Affiliations
- PMID: 21285522
- PMCID: PMC3026743
- DOI: 10.1172/JCI45691
Item in Clipboard
Review
Energy deficit in Huntington disease: why it matters
J Clin Invest.
2011 Feb.
Abstract
Huntington disease (HD) is an autosomal dominant neurodegenerative disease with complete penetrance. Although the understanding of the cellular mechanisms that drive neurodegeneration in HD and account for the characteristic pattern of neuronal vulnerability is incomplete, defects in energy metabolism, particularly mitochondrial function, represent a common thread in studies of HD pathogenesis in humans and animal models. Here we review the clinical, biochemical, and molecular evidence of an energy deficit in HD and discuss the mechanisms underlying mitochondrial and related alterations.
Figures
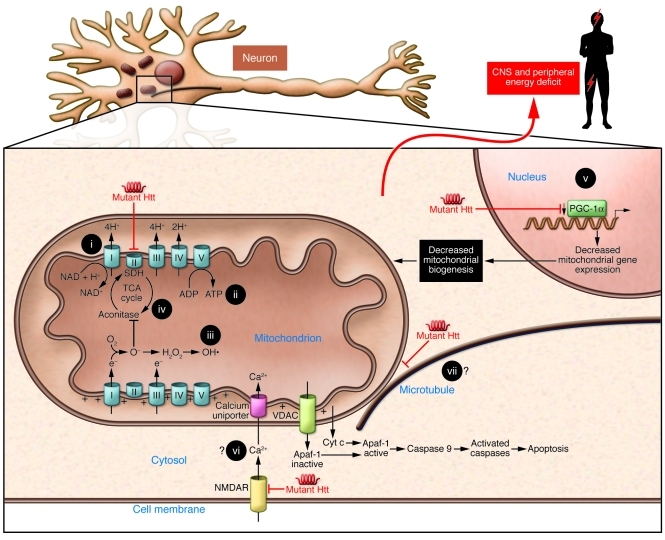
Such mechanisms would result in decreased mitochondrial biogenesis, oxidative stress, ATP deficit, increased apoptosis, and, ultimately, a central and peripheral energy deficit. Energy-related therapeutic approaches that have been used in preclinical models and/or HD patients include (i) coenzyme Q10, (ii) creatine, (iii) antioxidant therapies, (iv) anaplerotic therapies, and (v) PPAR agonists. Potential therapeutic targets are also shown, i.e., (vi) calcium homeostasis and (vii) mitochondrial transport. Apaf-1, apoptotic protease activating factor 1; NMDAR, N-Methyl-D-aspartic acid receptor; VDAC, voltage-dependent anion channel.
References
-
- Riley BE, Lang AE. Movement Disorders: Huntington’s Disease. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, eds.Neurology in Clinical Practice , 5th ed. Philadelphia, Pennsylvania, USA: Elsevier Publishing; 2008:1585–1586.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
